|
Peptide synthesis: chemical or enzymatic Fanny Guzmán Sonia
Barberis Andrés
Illanes* Financial support: This work was done within the framework of Project CYTED IV.22 Industrial Application of Proteolytic Enzymes from Higher Plants. Keywords: enzymatic synthesis, peptides, proteases, solid-phase synthesis.
Peptides are molecules of paramount importance in the fields of health care and nutrition. Several technologies for their production are now available, among which chemical and enzymatic synthesis are especially relevant. The present review pretends to establish a non-biased appreciation of the advantages, potentials, drawbacks and limitations of both technologies. Chemical synthesis is thoroughly reviewed and their potentials and limitations assessed, focusing on the different strategies and challenges for large-scale synthesis. Then, the enzymatic synthesis of peptides with proteolytic enzymes is reviewed considering medium, biocatalyst and substrate engineering, and recent advances and challenges in the field are analyzed. Even though chemical synthesis is the most mature technology for peptide synthesis, lack of specificity and environmental burden are severe drawbacks that can in principle be successfully overcame by enzyme biocatalysis. However, productivity of enzymatic synthesis is lower, costs of biocatalysts are usually high and no protocols exist for its validation and scale-up, representing challenges that are being actively confronted by intense research and development in this area. The combination of chemical and enzymatic synthesis is probably the way to go, since the good properties of each technology can be synergistically used in the context of one process objective.
Peptides are heteropolymers composed by amino acid residues linked by peptidic bonds between the carboxyl group of one amino acid residue and the α-amino group of the next one. The definition is rather vague in terms of chain length, peptides ranging from two to a few dozens residues. Its lower limit of molecular mass has been set rather arbitrarily in 6000 Da; molecules larger than that are considered proteins. Peptides are molecules of paramount importance in several fields, especially in health care and nutrition. The case of the hormone insulin (51 residues, 5773 Da) and the non-caloric sweetener aspartame (a dipeptide of aspartic acid and esterified phenylalanine) are relevant examples of those fields of application and the range of molecular size. Medium to small size peptides are, however, the most relevant for such applications. Different technologies are now available for the production of peptides and proteins: the extraction from natural sources (Hipkiss and Brownson, 2000), the production by recombinant DNA technology (Gill et al. 1996), the production in cell-free expression systems (Katzen et al. 2005), the production in transgenic animals (Wright et al. 1991) and plants (Cunningham and Porter, 1997), the production by chemical synthesis (Du Vigneaud et al. 1953; Merrifield, 1963) and by enzyme technology using proteolytic enzymes under conditions of displacement of the equilibrium of the reaction towards the formation of peptide bond (Feliú et al. 1995). The size of the molecule determines the technology most suitable for its production. Recombinant DNA technology is particularly suitable for the synthesis of large peptides and proteins, as illustrated by the case of insulin and other hormones (Walsh, 2005). Chemical synthesis is a viable technology for the production of small and medium size peptides ranging from about 5 to 80 residues (Kimmerlin and Seebach, 2005). Enzymatic synthesis is more restricted and has been hardly applied for the synthesis of peptides exceeding 10 residues. Its potential relies on the synthesis of very small peptides and, in fact, most of the cases reported correspond to dipeptides and tripeptides (Kumar and Bhalla, 2005). In this sense, the technologies for peptide production are not competitive with each other in most of the cases. The present review focuses on the chemical and enzymatic synthesis of peptides, aiming to establish a non-biased appreciation of their advantages, potentials, drawbacks and limitations. Chemical and enzymatic approaches do not exclude each other and a recent trend is the combination of both (Hou et al. 2005). The chemical route is often a better technological option than the biotechnological methods of recombinant DNA and biocatalysis for the synthesis of medium size peptides that comprise most of the pharmaceutically relevant molecules. It is also a fundamental tool for understanding the structure-function relationship in proteins and peptides, the discovery of novel therapeutic and diagnosis agents and the production of synthetic vaccines (Noya et al. 2003). Recently it has been used for the design of synthetic biocatalysts, which is a very promising area of research (Kaplan and DeGrado, 2004; Carrea et al. 2005). The synthesis of peptides was originally performed in solution. However, since the introduction of solid-phase synthesis by Merrifield (1986), this technology has gained more relevance (Stewart and Young, 1984) and significant advances have been made in the development of polymeric carriers and linkers, reversible protective groups (Goodman, 2002) and methods for the activation of covalent bond formation (Albericio, 2004), contributing in this way to the advancement of organic chemistry as a powerful tool for protein and peptide research. Synthesis of peptides in solution This technique has been used for the synthesis of small peptides composed by only a few amino acid residues. Its main advantage is that the intermediate products can be isolated and purified after each step of synthesis, deprotected and recombined to obtain larger peptides of the desired sequence. This technique is highly flexible with respect to the chemistry of coupling and the combination of the peptidic blocks. New strategies for synthesis in solution have been developed, going from the design of functional groups for the side chains and condensation of fragments for the synthesis of large molecules (Nishiuchi et al. 1998) to the use of new coupling reagents (Hiebl et al. 1999). Solid-phase synthesis of peptides Solid-phase peptide synthesis (SPPS) consists in the elongation of a peptidic chain anchored to a solid matrix by successive additions of amino acids which are linked by amide (peptide) bond formation between the carboxyl group of the incoming amino acid and the amino group of the amino acid previously bound to the matrix, until the peptide of the desired sequence and length has been synthesized (Nilsson et al. 2005). When Merrifield introduced the method of solid-phase synthesis in 1963, the scientific community reacted with skepticism: the synthesis in solution was at that time well established and in the new proposed system the purification of the peptide could only be done after cleavage, with the concomitant cleavage of most of the byproducts accumulated during the synthesis (Andersson et al. 2000). Despite these drawbacks, solid-phase synthesis has many advantages over the classical system in solution: the reaction can be automated and the problem of solubilization of the peptide no longer exists since it remains attached to the solid matrix. The strategy of synthesis (Fmoc or t-Boc), the nature of the solid carrier, the coupling reagents and the procedure of cleavage of the peptide from the solid matrix are the most relevant variables in SPPS. A general scheme of the stepwise SPPS is presented in Figure 1. The first step is the coupling of the C-terminal amino acid to the solid matrix. The Nα (A) group is then removed by treatment with trifluoroacetic acid (TFA) in the t-Boc strategy and with piperidine in the Fmoc strategy. The next (Nα protected) amino acid is coupled to the already synthesized peptide chain bound to the polymeric matrix and, once coupled, its Nα amino group is deprotected. This coupling-deprotection cycle is repeated until the desired amino acid sequence has been synthesized. Finally, the peptide-matrix complex is cleaved and side chain protecting groups are removed to yield the peptide with either a free acid or amide depending on the chemical nature of the functional group in the solid matrix. The cleavage reagent must remove the protecting groups of the side chains of the amino acids, which are stable at the conditions of Nα deprotection. Protection strategies. In the last few years, more than 250 protecting groups have been proposed as suitable for peptide synthesis (Goodman et al. 2001); however, a relatively small number of those is actually used because of the stringent requirements that a protecting group should meet, particularly with respect to the requirement of the preservation of other functionalities. Research and development in SPPS has conducted to two main schemes of protection, which are known as t-Boc/Bzl and Fmoc/tBu strategies (Chan and White, 2000). In t-Boc/Bzl, the t-Boc (tert-butoxycarbonyl) group is used for the protection of the Nα amino group and a benzyl or cyclohexyl for the side chains of several amino acids. In Fmoc/tBu, the Fmoc (9-fluorenyl methoxycarbonyl) group is used for the protection of the Nα amino group and the tert-butyl group for the side chains of several amino acids (Albericio, 2000). Protecting groups for the side chains commonly used in the t-Boc/Bzl and Fmoc/tBu strategies are listed in Table 1. Solid supports. Solid supports should meet several requirements: particles should be of conventional and uniform size, mechanically robust, easily filterable, chemically inert and chemically stable under the conditions of synthesis and highly accessible to the solvents allowing the penetration of the reagents and the enlargement of the peptide chain within its microstructure. They must not interact physically with the peptide chain being synthesized and should be capable of being functionalized by a starting group. Several polymeric supports are now available which can be derivatized with functional groups to produce a highly stable linkage to the peptide being synthesized (Barlos et al. 1991) and peptides with different functionalities in the terminal carboxyl group (i.e.: amide, acid, thioester) (Canne et al. 1999). Some examples are the p-methoxybenzhydrylamine (MBHA), 4-hydroxymethyl-phenylacetamidomethyl (PAM) and hydroxymethyl functionalized resins used for t-Boc/Bzl, and the 4-(2',4'-dimethoxyphenyl-aminomethyl)-phenoxymethyl-polystyrene (Rink), 2-chlorotrityl chloride, and diphenyldiazomethane functionalized resins used for Fmoc/tBu. Coupling reagents. Several reagents that activate the carboxyl groups of the amino acids are used for the coupling reaction. The most used now are uronium and phosphonium salts and hydroxybenzotriazole (HOBT) because of the high reactivity, high coupling yield and higher specificity than obtained in conventional systems, such as DCC/HOBT (dicyclohexyl-carbodiimide/1-hydroxybenzotriazol), HOBT derivatives and symmetric anhydrides (Miranda and Alewood, 2000). Chaotropic salts (CuLi, NaClO4, KSCN) and mixtures of solvents, such as N,N dimethylformamide, trifluoroethanol, dimethylacetamide and N-methylpirrolidone, have been used to improve the efficiency of coupling and the elongation of the peptide chain in tough sequences. Cleavage from the solid support. Once the peptide synthesis of the desired sequence is finished, the protecting groups of the side chains are removed and the peptide freed from the support. In the t-Boc/Bzl strategy, the most popular method is the one developed by Tam et al. (1983). Deprotection is carried out with strong acids that may lead to unwanted secondary reactions of alkylation or acylation in certain amino acids that are promoted by the leaving protecting groups. To avoid such reactions, combinations of solvents acting as nucleophiles and acids that allow the process of deprotection have been pursued for decades. Different is the case of the Fmoc/tBu strategy, in which simpler solvents as TFA in combination with triisopropylsilane can be used. The main SPPS strategies are sequential synthesis, convergent synthesis and chemical ligation. Sequential synthesis involves the stepwise addition of amino acids until the desired sequence is synthesized. Convergent synthesis involves the independent synthesis of peptide sequences that are then linked by condensation in liquid phase. In chemical ligation those fragments are linked by chemoselective reactions involving thioether (Lu and Tam, 2005), oxime (Nardin et al. 1998), hydrazone and thiazolidine (Tam et al. 1995) linkages. Sequential. Sequential synthesis was already depicted in Figure 1. This strategy is used for the synthesis of small to medium size peptides having up to 50 residues. However, larger size polypeptides can be constructed using sequential synthesis by the technique of cysteine polymerization, the construction of dendrimers using lysine matrices, or the construction of Template-Assembled Synthetic Protein (TASP) (Tuchscherer and Mutter, 1996; Banfi et al. 2004). Cysteine polymerization is performed by locating cysteine residues at the amino and carboxy terminals of the peptide. Cysteine has a sulfhydryl group prone to oxidation to form disulfide bridges so that the peptide can be polymerized both at the amino and carboxy terminals to yield polypeptides of high molecular weight. This technique produces a wide range of oxidized peptidic species ranging from the cyclic monomer and dimer to high molecular weight polypeptides, as shown in Figure 2. Cysteine polymerization was used for the construction of the first chemically synthesized anti-malaria vaccine, Spf-66 (Patarroyo et al. 1988; Amador et al. 1992). Cysteine chain polymer peptides are often the best presentation of a peptidic vaccine in terms of immunogenicity (Patarroyo et al. 2002). The problem is that it is not possible to tightly control the degree of polymerization to obtain a single peptide species, so that different species are produced whose distribution is not necessarily reproducible from one batch to another. This drawback can be important for the construction of human vaccines and for the validation of the process. In fact, proper control of SPPS by cysteine polymerization still represents a challenge. High molecular weight peptides can be conveniently synthesized by the t-Boc/Bzl strategy using in-situ neutralization (Miranda and Alewood, 1999; Taylor et al. 2005), which cannot be easily done by Fmoc/tBu because of the steric hindrances in the deprotection stage of the Fmoc group as the peptide chain grows (Tickler et al. 2001). Another option for obtaining high molecular weight peptides is the construction of dendrimers, which are highly ordered and branched peptides that contain in their core a matrix of lysine residues to whose amino groups several copies of the peptide sequence of interest can be linked. In this way a peptidic macromolecule with several copies of the desired sequence can be synthesized. A type of dendrimer, known as multiple antigen peptide system (MAP), composed of a lysine matrix and an external zone of antigenic peptides is illustrated in Figure 3 (Tam, 1988). One of the problems of this technique is that truncated or incomplete sequences of the peptide may be synthesized in some of the branches of the structure. Dendrimers known as double dimer constructs (DDC) have been reported by Calvo et al. (1999). The construction of a DDC occurs in two steps: initially a peptide is constructed with two arms containing a cysteine at each carboxy terminal; then, this dimer is purified and subsequently oxidized forming a tetramer as shown in Figure 4. DDC type tetramers have been used as immunogens in the study of possible candidates for anti-malaria vaccines, being this presentation more immunogenic than the one based on cysteine polymers in the experimental model with Aotus monkeys (Rivera et al. 2002). Compared with MAPs, DDCs produce higher yields of synthesis of branched peptides and its purification is simpler (Chávez et al. 2001; Rivera et al. 2002). Its main limitation is that this system is not applicable to dendrimers with copies of antigens whose sequences have conformational restrictions. A dendrimeric architecture containing eight variable positions connected by three successive branching diamino acid units was recently used to develop a library of potentially catalytic peptides (Clouet et al. 2004). Combinatorial libraries have been also developed for peptide enzyme mimics (Kofoed et al. 2006). Convergent. In convergent synthesis, peptides (up to 50 residues) are separately produced by sequential synthesis and then linked in solution or in solid phase to obtain the desired high molecular weight peptide or protein. A schematic representation of convergent synthesis is presented in Figure 5. The advantage of convergent synthesis is that each peptide fragment is purified and characterized before being linked. In this way, side reactions are minimized during synthesis. Depending on the protective groups and the functional group linked to the support, it is possible to use some orthogonal protection scheme in which two or more types of groups are involved that are removed by different mechanisms (Zhang and Tam, 1999). An example of orthogonality in convergent synthesis is the combination of t-Boc/Bzl and Fmoc/tBu using supports whose linkage to the peptide can be either labile to acid, base, palladium or photolabile. These combinations allow the selective protection or deprotection of the reactive groups to avoid side reactions and direct the synthesis. However, convergent synthesis has some drawbacks: the solubility of the protected fragments in the aqueous solvents used in the purification by HPLC and in the organic solvents used in the coupling reactions is usually low, reaction rates for the coupling of fragments are substantially lower than for the activated amino acid species in the conventional stepwise synthesis and, finally, the C terminal of each peptide fragment can be racemized during coupling. Some of these problems have been circumvented by using mixtures of solvents to increase the solubility, by using prolonged reaction times to increase the efficiency of coupling and by using glycine or proline in the C terminal to avoid the problem of racemization. In this way, despite the technical problems yet to be solved, convergent synthesis represents the best option for the chemical synthesis of large peptides and proteins. A variety of large peptides have been successfully produced by convergent synthesis, such as P41icf (Chiva et al. 2003) and T-20 (Bray, 2003). New generation globular polyproline dendrimers have bee synthesized recently by convergent SPPS which are relevant structures for the delivery of peptide drugs (Fillon et al. 2005; Sanclimens et al. 2005). Chemical ligation. Chemical ligation is a particularly appealing strategy for the chemical synthesis of large peptides and proteins (Baca et al. 1995; Yan and Dawson, 2001). It is based on the chemical linkage of short unprotected peptides which are easy to handle because of its high solubility in the solvents used for synthesis. These peptides are functionalized with groups that react chemoselectively with only one group of the acceptor peptide preserving the integrity of the unprotected side chains. Many proteins and peptides of biological interest have been synthesized by chemical ligation using a variety of ligands with the formation of thioester, oxime, disulfide, thiazolidine and peptide bonds. Initially, the main problem was the incorporation in the peptide sequence of non-natural (non-peptide) bonds; however, there are several examples of these non-natural structures that maintain the activity of the natural peptide. Based on the original principle of chemical ligation, Dawson et al. (1994) developed a method that allows the linkage of unprotected peptide segments by thioester bond that suffers a spontaneous rearrangement to produce a peptide bond. This strategy is termed native chemical ligation (Figure 6) and several peptides and proteins have been synthesized by this method without the introduction of non-peptidic bonds (Dawson and Kent, 2000). The method involves a thioester mediated reaction in aqueous solution at neutral pH of two unprotected peptide segments resulting in the ligation of the terminal cysteine of one peptide segment to the other peptide segment via a native amide bond. Some typical examples of this methodology are the synthesis of phospholipase A (124 residues with 14 cysteine residues and 7 disulfide bridges), the synthesis of the microprotein S (Hackeng et al. 2000) and the total chemical synthesis of the H-Ras proteins of the c-Raf-1 effector having 166 residues (Becker et al. 2003). A review on the synthesis of peptides by chemical ligation has been recently published (Johnson and Kent, 2006). Experimental procedures. SPPS can be performed in different ways. There are manual and automated systems available for small and large scale synthesis of only one peptide or several peptides at the time (multiple peptide synthesis). All operations in SPPS, namely coupling, deprotection and final removal, are conducted in the same recipient so that several washing steps have to be considered; reagents are used in large excess to speed-up the reaction and drive it to completion. Manual synthesis of individual peptides can be performed in syringes of different sizes provided with a bottom sintered-glass or plastic filter. Multiple peptide synthesis at the micromolar level can be conducted in functionalized cellulose, polypropylene or polyvynilidene difluoride membranes, according to the spot-synthesis methodology developed by Frank (2002). Fmoc/tBu and t-Boc/Bzl multiple peptide synthesis at the millimolar level can be performed using the tea-bag methodology developed by Houghten (1985), in which up to 400 peptides of 20 residues or less can be synthesized at the same time. The tea-bag system of SPPS has been successfully employed to construct peptide libraries (Houghten et al. 2000). Several systems for the automated t-Boc/Bzl and Fmoc/tBu SPPS, going from 1.5 mg to 5 kg scale, are now available from Applied Biosystems, Shimadzu, Advanced ChemTech, Protein Tecnologies, Abimed and Zinsser Analytic, among others (Chan and White, 2000). These systems allow a tight control of the conditions of synthesis, which is important for the reproducibility of results and the validation of the process. However, automated SPPS is expensive and a thorough economic evaluation should be done in each case before adopting it. During the process of peptide synthesis, several analyses must be performed at intermediate stages but mostly to the final product. During synthesis, the determination of free amino groups is essential in the stages of coupling and Nα deprotection in the case of Fmoc strategy. The most common assay for free amino groups is the ninhydrine test because of its sensitivity and reproducibility, but it is a destructive test so that samples have to be removed from the product being synthesized (Sarin et al. 1981). Non destructive tests are also used. The bromophenol blue (Krchñak et al. 1988) and picric acid (Gisin and Merrifield, 1972) tests are prominent among non-destructive tests, but they are not so commonly used because of their lesser sensitivity and reproducibility; in the case of picric acid test a trained eye is required to assess the difference in yellow tones. Reverse-phase high performance liquid chromatography (HPLC), using C-18 or C-8 columns, is the most used procedure for analysis and purification of peptides. Separation is based on hydrophobicity and mobile phase gradients are used with polar solvents such as acetonitrile. When peptides are synthesized as cysteine polymers, the molecular mass of the product can be determined by size-exclusion HPLC (Santovena et al. 2002) or capillary electrophoresis (Rosas et al. 2001). The determination of the molecular mass of the peptide is fundamental for its proper identification and mass spectrometry (MS) is the most powerful analytical technology available, which can also give information on structural modifications of a particular peptide (i.e. alkylations, formation of aspartiimides) that occur during cleavage of the peptide in SPSS. It is extremely rapid and hundreds of samples can be analyzed sequentially over extremely small amounts of peptides (Kalia and Gupta, 2005). MS can be divided into two main categories depending on the method of ionization, which are termed matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI). MALDI is the most powerful technique nowadays for molecular mass determination of peptides, since it produces no fragmentation. Most used matrices for MALDI are 2-5 dihydroxybenzoic acid (DHB), 3-5 dimethoxy-4-hydroxy cinnamic acid (sinapinic acid, SA) and α-cyano-4-hydroxycinnamic acid (CCA) (Moore, 1997). MALDI is an extremely sensitive method: samples ranging from 5-10 pmoles/µL can be assayed and only one µL is required. ESI shares the good properties of MALDI but has some additional restrictions since only rather polar molecules can be analyzed (Kalia and Gupta, 2005). Both MALDI and ESI are less sensitive for high molecular weight peptides (over 30000 Da). The method of choice will depend on the type of peptide, although both systems deliver in most cases the same type of information (Ambulos et al. 2000). The precise quantification of the amount of peptide synthesized is determined through amino acid analysis by HPLC after peptide hydrolysis in acid medium considering those acid-resistant amino acids. Although amino acid analysis is usually performed after cleavage from the resin, it can also be conducted to the peptide still attached to the resin (Ambulos et al. 2000). The quantification of cysteinic SH groups in the peptide is performed according to the method of Ellman (1958). The determination of peptide structure can be done by circular dichroism (CD) (Brahms and Brahms, 1980) and nuclear magnetic resonance (NMR) (Rance et al. 1983). Secondary structure determination by CD can be done with only 20 µg of peptide on a 0.1 mL cell. The analysis will determine if the peptide has a secondary structure and if the structure is α-helix (one maximum at 193 nm and two minima at 208 and 222 nm), or β-sheet (one maximum at 198 nm and one minimum at 217 nm). If the peptide has no secondary structure, only one maximum will be obtained at 211 nm. Other relevant structures in peptides are β-turns, which can be mistaken as no structure, since they yield a similar spectrum (Guzmán et al. 2002). NMR requires from 7 to 10 mg of peptide per sample, but in addition it provides information about the amino acid residues involved in each structure (Bermúdez et al. 2003). The application of native peptides for pharmacological use may have some restrictions because of the degradation by endogenous proteases, hepatic clearance, undesired side effects by interaction with different receptors, and low membrane permeability due to their hydrophilic character (Sewald and Jakubke, 2002). Chemically modified peptides with improved bioavailability and metabolic stability may be directly used as drugs and many efforts have been made to develop peptide-based, pharmacologically active compounds. The most straightforward approach for peptide modification is to introduce changes into the side chains of single amino acids. The introduction of such functional groups that do not occur naturally in peptides restricts its conformational flexibility and enhances its metabolic stability. Other option is the modification of the peptide chain (backbone), which can be accomplished in different ways: its exchange by an amide analogue, the alkylation of the NH group of the peptide bond (Ahn et al. 2002), the reduction of the carbonyl group of the peptide bond (Lozano et al. 2004), or the exchange of the NH group of a peptide bond by an oxygen atom (depsipeptide) (Kuisle et al. 1999; Albericio et al. 2005; Cruz et al. 2006) a sulfur atom (thioester) (Vabeno et al. 2004) or a CH2 group (ketomethylene isostere) (Martin et al. 2001). Notable among these modified peptides are depsipeptides that are compounds having sequences of amino and hydroxy carboxylic acid residues regularly alternating; they can also form cyclic structures named cyclodepsipeptides, where the residues are connected in a ring. There is great interest in this class of products due to their wide range of biological properties such as immunosuppressant, antibiotic, antifungi, antiinflammatory and antitumoral activities (Hamel and Covell, 2002; Sarabia et al. 2004). Another approach is the construction of constrained structure peptidomimetics in which a region of a defined structure in the natural peptide is synthesized and bound by its amino and carboxy terminals through a non protein ligand (Ehrlich et al. 1996; Lioy et al. 2001). In this way, better recognition by antibodies can be obtained (Calvo et al. 2003). Such peptidomimetics have also been produced to inhibit protein-protein interaction (Yin et al. 2006). Peptides can also be glycosylated. In fact, many proteins and peptides of pharmacological relevance are naturally glycosylated and in such cases the carbohydrate portion of the moiety can be essential for recognition and improvement of clearance rate and other pharmacodynamic parameters. In the chemical synthesis of glycopeptides, glycosylation can be obtained either by using glycosylated amino acids or by conjugation of the final carbohydrate unit to the full-length peptide (Von Moos and Ben, 2005). Several other reactions of modification of peptides (i.e. with biotin, fluorescein, rhodamine) at its amino terminal are useful as markers for conducting in vitro kinetic studies (Chan and White, 2000). The synthesis of β-peptides (peptides constructed from non-natural β-amino acids) is a new promising area of peptide research. Short chains of β-amino acids usually form more stable secondary structures than the natural α counterparts and have proven to be endowed with interesting biological properties, for instance: inhibition of cholesterol and fat intestinal absorption (Werder et al. 1999). These non-natural peptides are resistant to enzymatic degradation which can be a relevant characteristic for their therapeutic application (Schreiber et al. 2002). Small β-peptides can be synthesized using conventional Fmoc/tBu SPPS strategy (Guichard and Seebach, 1997), but the protocols have to be modified for longer peptides because of the high folding propensity of β-peptides, which increases coupling times and prevents complete deprotection. However, the replacement of piperidine by stronger bases has circumvented the problem of incomplete deprotection in Fmoc/tBu solid phase synthesis of β-peptides (Schreiber and Seebach, 2000) and longer β-peptides and mixed type α-β-peptides have been successfully synthesized by chemical ligation (Kimmerlin et al. 2002). β-amino acids are, however, very expensive building blocks so that the usual practice in SPPS of using excess reagents has to be avoided. This problem has not been fully addressed and remains to be solved from a technological point of view. Methodologies for β-peptide synthesis have been recently reviewed (Kimmerlin and Seebach, 2005). Large scale chemical synthesis of peptides More than 40 therapeutic peptides are now in the market, which represents a considerable increase, since less than 10 were in the market by 1990. A much higher number of therapeutic peptides are in the different phases of approval. The therapeutic application of peptides has an enormous potential, which has been enhanced by the advances in the fields of formulation and administration of pharmaceuticals. Among the technologies available for the large scale synthesis of peptides, chemical synthesis is the most developed and SPPS can be considered now a mature technology. SPPS can be used sometimes in combination with synthesis in solution (hybrid synthesis). Even though SPPS can be troublesome in practice for peptides of more than 10 residues, in some cases peptides of about 50 residues have been synthesized successfully by this system (Patarroyo et al. 1988) and several peptides from 30 to 50 residues synthesized by SPPS are now in the market (Bruckdorfer et al. 2004). Commonly, peptides of less than 30 residues are produced entirely by solid phase sequential synthesis (Lloyd-Williams and Giralt, 2000), while larger peptides (up to 60 residues) must be produced by convergent synthesis in which protected peptide fragments are synthesized by SPPS and then combined by liquid phase synthesis (Riniker et al. 1993; Barlos and Gatos, 1999). Proteins are preferably produced by chemoselective ligation in which all the unprotected linked fragments have been previously synthesized by SPPS. The development and commercialization of acid labile resins (Rink, 1987; Bray, 2003) has allowed the synthesis of large peptides by hybrid synthesis. Small fragments with protected side chains are synthesized over the acid-labile resins using Fmoc protected amino acids; these fragments are removed from the resin with 1% TFA solution. The fragments that contain the whole peptide or protein sequence are condensed in solution to yield a product with protected side chains which is then deprotected and purified to complete the synthesis of the desired product. Independent of the SPPS system chosen, peptide synthesis is a multi-step process where the number of operations required increases proportionally to the number of amino acid residues of the peptide. This in turn produces a decrease in yield, which is exponential with the number of steps involved in the synthesis of the peptide. In fact, if Ri is the yield of recovery in step i, then the global yield of recovery (R) of a synthesis of N steps is:
The
synthesis of a peptide of 10 to 20 amino acids involves 20 to 50 operations.
If an average yield of recovery of 90% is assumed for each amino acid
linked, the total yield of recovery would be within the range of 35%
(0.910 = 0.35) and 12% (0.920 =
0.12). Even if the average yield of recovery could be increased to
98%, the total yield of recovery would be within 82% (0.9810
= 0.82) and 67% (0.9820 = 0.67). This simple exercise highlights
the importance of improving the coupling efficiencies, which is a
key aspect for the success of a process for peptide synthesis at industrial
scale. If one takes into consideration that in the production of The synthesis of sophisticated peptides for therapeutic use is first conducted at the milligram scale for performing biological studies and elucidating structure-activity relationships. Even though yields of recovery can be quite low at this stage, they can be good enough to conduct such studies. However, the identification of the peptide as a candidate for clinical studies demands the synthesis of large amounts of product. When the therapeutic dose has been established in Phase II, the scale of production will be higher and a scale-up strategy for the process will be necessary. At this point, the cost-benefit analysis of the process of synthesis becomes relevant. The strategy should satisfy not only the requirements for the licensing of the product but also comply with the regulatory requirements and validation of the process, which implies the establishment of the chemical and biological equivalence with the product synthesized at the small scale. In this context, the amino acid protection systems should be similar, and ideally identical, in both stages of development. The selection of the most effective route for synthesis at large scale represents a challenge and all efforts should be directed towards the design of the procedure of synthesis that renders the higher productivities and yields. When the process is scaled up to production levels (kilograms or tons, depending on the product), criteria that are not necessarily relevant at small scale become relevant. Production costs, purity of the product, security and environmental impact of the process are now issues of major concern. Costs of reagents are usually high; therefore, the use of large excess, which is common practice in the early stages of development, is inadmissible at large scale, where it is highly desirable to use reagents as close to stoichiometry as possible. In fact, large amounts of reagents cannot be wasted just to ensure the completion of the reaction. Aside from costs, environmental burden is also a matter of concern because of the surplus effluent produced. Therefore, the kinetics of the reactions should be studied in greater detail when scaling up to reduce the costs associated with reagents and waste treatment after reaction (Andersson et al. 2000; Bruckdorfer et al. 2004; Patarroyo and Guzmán, 2004). Some relevant examples of peptides produced at large scale and the production system used are shown in Table 2. The final goal of a process for the large scale synthesis of a peptide is to establish its technological and economic viability and to satisfy the requirements of the regulatory agencies in terms of product quality standards and process validation. A system of GMP (good manufacturing practices) must be established to ensure that the production process is highly reproducible and the final product is delivered with a consistent quality (WHO, 1999). Process validation should consider reproducibility in terms of yields of intermediate and final products and consistency in the profile of impurities of the product (Andersson et al. 2000). This is a complex task in the case of the synthesis of peptides because of the complexity and the number of operations involved in the production process. The stringent requirements for validation, despite the high cost and long time required, has to be appreciated as the adequate way to ensure the higher standards of quality and safety required by the final consumer, which is the recipient patient. Enzymes are the biological catalysts responsible for cell metabolism. As such, they must perform well at the mild conditions required for cell functioning. To become process biocatalysts they must be robust enough to withstand the harsh conditions of an industrial process, which usually implies the modification of the enzyme to produce a stable biocatalyst (Illanes, 1999). Proteolytic enzymes comprise a group of hydrolases that is the most relevant in technological terms, sharing about one half of the world market of enzymes, with annual sales of about US$ 3 billion (Chellapan et al. 2006). Microbial and plant proteases are the most relevant and have been widely utilized in medicine and in different industrial processes for decades (Wiseman, 1980; Hultin, 1983; Klein and Langer, 1986; Van Brunt, 1987). Microbial proteases are the most important in terms of market-share because of the advantages of their intensive production. However, plant and some animal proteases are still relevant for certain medical and industrial applications. Microbial and plant proteases have been traditionally used in degradation processes and are available at low cost; however, new sources of proteases are required to produce more potent and specific enzymes. New proteases from endogenous plant species have been characterized and proved their efficacy for performing hydrolytic and synthetic reactions (Arribére et al. 1998; Arribére et al. 1999; Priolo et al. 2000; Obregón et al. 2001). New sources of proteases are continuously being reported, especially from exotic organisms that thrive in extreme environments, being their proteases abnormally stable and/or active at such extreme conditions. As an example, the thermophilic and alkalophilic proteases from the salt tolerant marine fungus Engyodontum album has been recently reported and characterized (Chellapan et al. 2006). Proteases are active at mild conditions, with pH optima in the range of 6 to 8; they are robust and stable, do not require stoichiometric cofactors and are also highly stereo and regioselective (Bordusa, 2002). These properties are quite relevant to use them as catalysts in organic synthesis. This is possible because proteases can not only catalyze the cleavage of peptide bonds but also their formation (Capellas et al. 1997; Björup et al. 1999; So et al. 2000), as well as other reactions of relevance for organic synthesis, for instance: the regiospecific hydrolysis of esters and the kinetic resolution of racemic mixtures (Khmelnitsky et al. 1997; Carrea and Riva, 2000; Bordusa, 2002). Subtilisin, chymotrypsin, trypsin and papain have been widely used proteases in the chemical synthesis of peptides. Proteolytic enzymes: classification and catalytic mechanisms Proteases are among the best studied enzymes in terms of structure-function relationship (Barret et al. 1998; Priolo et al. 2001; Drauz and Waldmann, 2002). The proteases that are used for peptide synthesis are selected on the basis of their specificity against amino acid residues on either side of the splitting point (Kumar and Bhalla, 2005) and include the majority of the commercially available endo and exoproteases. The broad specificity of proteases restricts their application in peptide synthesis, since the peptide product that accumulates during the reaction can be attacked by the proteases simultaneously with the reaction of synthesis (Schellenberger et al. 1991). There are five families of proteases in which serine, threonine, cysteine, aspartic or metallic groups play a primary catalytic role. Serine, cysteine and threonine proteases are quite different from aspartic and metalloproteases. In the first three groups, the nucleophile in the catalytic center is part of an amino acid residue, while in the second two groups the nucleophile is an activated water molecule. In cysteine proteases the nucleophile is a sulfhydryl group and the catalytic mechanism is similar to the serine proteases in which the proton donor is a histidine residue (Figure 7 and Figure 8). Besides the hydrolysis and formation of peptide bonds between amino acids, proteases can catalyze other types of reactions as well, such as esterification and transesterification in the resolution of racemic alcohols and carboxylic acids and the stereoselective acylation of meso and prochiral diols (Broos et al. 1995; Khmelnitsky et al. 1997; Liu and Tam, 2001), the synthesis of glycoconjugates (Wong et al. 1993; Bordusa, 2002) and the kinetic resolution of racemic mixtures, even though in this case lipases and esterases are more useful for the resolution of non amino acidic derivatives (Kirchner et al. 1985; Bornscheuer and Kazlauskas, 1999; Pogorevc and Faber, 2000; Roberts, 2001). On the other hand, non-proteolytic enzymes can also be used in connection with peptides, as illustrated by the kinetic resolution of N-phenylacetyl-DL-tert-leucine in the production of the non-proteinogenic chiral amino acid L-tert-leucine recently reported with penicillin G acylase from Kluyvera citrophila (Liu et al. 2006). Proteases in peptide synthesis Biocatalysis in non-conventional (non-aqueous) media has expanded the spectrum of application of proteases to those reactions that cannot proceed effectively in aqueous environments, this is, the synthesis of peptide bonds instead of their hydrolysis (Illanes and Barberis, 1994; Quiroga et al. 2000a). These reaction media, that include organic solvents (Clark, 2004; Gupta and Roy, 2004), supercritical fluids (Kamat et al. 1992; Mesiano et al. 1999), eutectic mixtures (Gill and Vulfson, 1994), solid-state (Halling et al. 1995; Erbeldinger et al. 1998) and, more recently, ionic liquids (Park and Kazlauskas, 2003; van Rantwijk et al. 2003; Lou et al. 2004; Machado and Saraiva, 2005) offer other potential advantages as well: the possibility of using poorly water soluble substrates; the modification of the equilibrium of reaction as a consequence of the alteration of the partition coefficients of substrates and products in the case of biphasic systems; the reduction of inhibitory effects by substrates and products; the easiness of biocatalyst and product recovery; the increase in the thermostability of the biocatalyst and, in some cases, the variation in substrate specificity and the increase in the stereo and enantiospecificity in the resolution of racemic mixtures (Kawashiro et al. 1997; Klibanov 2001). As compared to chemical synthesis, a most important advantage of biocatalysis is the specificity of the reaction, which reduces the requirement of side-chain protection. Enzymatically synthesized small peptides (usually di or tripetides) are being used successfully for human and animal nutrition and also as pharmaceuticals and agrochemicals. Some relevant examples are the synthesis of the leading non-caloric sweetener aspartame, the lysine sweet peptide, kyotorphin, angiotensin, enkephalin and dynorphin (Kullman, 1979; Takai et al. 1981; Kullman, 1982; Oyama et al. 1987; Aso, 1989; Clapés et al. 1989; Nakanishi et al. 1990), and some nutritional dipeptides and tripeptides (Kimura et al. 1990b, Kimura et al. 1990c; Monter et al. 1991). Some small peptides have been synthesized at commercial scale in continuously operated enzyme reactors (Herrmann et al. 1991; Serralheiro et al. 1994). Kimura et al. (1990b) have proposed various kinds of reactors to synthesize peptides from essential amino acids using papain, α-chymotripsin, and thermolysin. Telios Pharmaceutical Co. has explored the enzymatic synthesis of the tripeptide Arg-Gly-Asp as a new drug for the healing of heavy burns and dermal ulcer (Chen et al. 1998). Several other examples of enzymatically synthesized peptides have been reported in the last decade (Monter et al 1991; Murakami and Hirata, 1997; Murphy and Fagain, 1997; Sergeeva et al. 1997; Zaks and Dodds, 1997, Liu et al. 2002). We have synthesized biologically active small peptides using proteases isolated from the latex of plants in biphasic reaction systems (Barberis et al. 2002). Synthesis under thermodynamic control. Peptide synthesis by proteases can proceed by two mechanisms: thermodynamic and kinetic control (Kumar and Bhalla, 2005), which will be described in this and the following section respectively. The thermodynamically controlled synthesis of peptides (TCS) with proteases represents the reverse of the hydrolytic breakage of peptide bond catalyzed by those enzymes, as shown in the scheme (Jakubke et al. 1985):
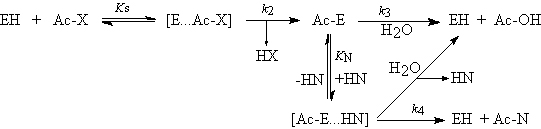
were Kion is the equilibrium constant of ionization and Kcon is the equilibrium constant of conversion. Proteases, as any catalyst, do not alter the equilibrium of the reaction but merely increases the rate to attain it. According to the principle of microscopic reversibility, both the formation and the hydrolysis of the peptide bond proceed by the same mechanism and through the same intermediate. The formation of the acyl intermediate from a carboxylic acid is a very slow reaction and represents the rate limiting step in TCS (Sakina et al. 1988). Main issues of TCS are the use of an acyl donor with the free carboxylic group and the possibility of using any type of proteases, independent of their catalytic mechanism. Their main drawbacks are the low reaction rates and product yield (determined by the equilibrium constant of the reaction) attainable, the high amount of enzyme biocatalyst often required and the need of precise reaction conditions to displace the equilibrium towards synthesis (Bordusa, 2002). This in practice can produce severe compromises with enzyme activity and stability. From the above scheme, it is apparent that the equilibrium will be displaced to hydrolysis in an aqueous medium. The displacement of equilibrium towards peptide bond formation can be attained by manipulation of the equilibrium of ionization (i.e. pH change) and the equilibrium of the reaction (i.e. by product precipitation or by modification of medium composition) (Homandberg et al. 1978; Jakubke et al. 1985). The addition of organic cosolvents and the use of aqueous-organic biphasic systems are good strategies to displace the equilibrium towards synthesis. The presence of organic solvents will reduce the activity of water in the reaction medium, which will favour the equilibrium, and will also reduce the dielectric constant of the medium, which will reduce the acidity of the carboxylic group of the acyl donor and so it will increase the equilibrium constant Kion promoting the reaction of synthesis. The use of cosolvents is a rather simple strategy but high concentrations of cosolvents are usually detrimental for enzyme activity (Jakubke et al. 1985). In biphasic systems, the partition of the peptide products from the aqueous phase that contains the enzyme to the organic phase drives the equilibrium towards synthesis, with the additional benefit that the product is no longer subjected to hydrolysis. However, reaction rates in biphasic systems are low because of limitation by substrate diffusion; besides, proteases tend to denature in the water-solvent interphase (Halling, 1994). The use of neat hydrophobic solvents with very low water content can in principle be effective for peptide synthesis by reducing the backward hydrolytic reaction. However, proteases may exhibit very low activity and reduced stability, and substrates and products can be poorly soluble in this kind of media. Replacing the essential water by other hydrogen bond forming solvents can increase oligopeptide solubility. In summary, each process of TCS requires of a study to determine which of the process alternatives (water-cosolvent medium, biphasic medium or hydrophobic solvents with low amount of water) is the best. The choice will depend on the characteristics of the reaction, the robustness of the biocatalyst and also on the properties of substrates and products. Synthesis under kinetic control. The kinetically controlled synthesis of peptides (KCS) with proteases can be represented by the following scheme (Bordusa, 2002):
where EH is the free enzyme; Ac-X is the acyl donor substrate; [E...Ac-X] is the Michaelis-Menten acyl-enzyme complex; HX is the released group; Ac-E is the acyl-enzyme intermediate, HN is the acceptor substrate (nucleophile), Ac-N is the product of synthesis (peptide) and Ac-OH is the product of hydrolysis of the acyl donor. As shown in the above scheme, the acyl donor, that needs to be activated in the form of an ester, an amide or a nitrile, first binds to the enzyme to form a tetrahedric enzyme-substrate complex [E...Ac-X] that collapses to form the covalent acyl-enzyme intermediate [Ac-E]. This intermediate can be nucleophilically attacked by water and by the nucleophile (HN), which can be an amine, an alcohol or a thiol that will compete with water for the deacylation reaction. The success of the reaction of synthesis will depend on the kinetics of these nucleophilic reactions; this is why this strategy is termed kinetically controlled. As opposite to TCS, only serine or cysteine proteases can be used to perform KCS, because the enzyme acts in this case as a transferase and catalyzes the transference of an acyl group from the acyl donor to the amino acid nucleophile through the formation of an acyl-enzyme intermediate. Generally KCS proceeds faster and requires lower enzyme to substrate ratios than TCS because the acyl donor is now in the form of an activated carboxylic acid (Bordusa, 2002). Papain, thermolysin, trypsin and α-chymotrypsin are the most used enzymes in KCS (Mitin et al. 1984; Ooshima et al. 1985; Nakanishi and Matsuno, 1986; Reslow et al. 1987; Nakanishi and Matsuno, 1988; Mitin et al. 1997; Björup et al. 1998; Fité et al. 1998; Björup et al. 1999). It is desirable that the peptide product be removed from the reaction medium to avoid unwanted secondary hydrolysis. The yield of peptide product will depend on the apparent ratio of transferase to hydrolase rate constants (KT/KH)app and the rate at which the peptide product is hydrolyzed. Protease used in KCS have values of (KT/KH)app in the range of 102-104 (Kasche, 1996). Biphasic systems are not adequate to perform KCS since in this case, the neutral esters commonly used as acyl donors partition poorly into the aqueous phase and therefore the concentration in that phase, where the enzymatic reaction occurs, is low. However, the synthesis of N-α-CBZ-Ala-Phe.OMe was successfully accomplished in a biphasic system composed by Tris-HCl buffer (pH 8.5) and ethylacetate (50%), using a crude protease from the latex of Araujia hortorum Fourn (Quiroga et al. 2000b). In this case, the favourable product partition coefficient to the organic phase allowed its recovery from this phase and increased yield; besides, the biocatalyst exhibited high specificity and selectivity for the synthesis of the dipeptide without significant polymerization. As in TCS, the decrease of water activity, by using an organic cosolvent, favours synthesis in KCS by reducing the hydrolysis of the acyl-enzyme intermediate and the final product, but, again, the reaction medium can be harmful to the enzyme (Cassells and Halling, 1990; Barberis et al. 2002). Mutant enzymes have been used in the KCS of peptides in alkaline medium containing organic cosolvents where the parent enzymes were denatured, obtaining high conversion yields (West et al. 1990; Wong et al. 1990). Temperature, pH, substrates concentrations and enzyme to substrate ratio are parameters that will affect KCS (Chen et al. 1994). An increase in nucleophile concentration will increase the rate of nucleophilic attack to the acyl-enzyme intermediate (Jakubke, 1987); an increase in pH will also be beneficial since it will increase the pK of the nucleophile whose only reactive form is the neutral species (Barberis et al. 2002; Bordusa, 2002). As opposed to TCS, the enzyme itself will determine the efficiency of the peptide synthesis in KCS. While the reaction rate is mainly determined by the specificity of the enzyme for the acyl donor, a specific linkage of the nucleophile to the S subsite of the protease is essential for a high yield of synthesis (Schechter and Berger, 1967). Since the specificity for the acyl donor and the nucleophile linkage are individual parameters for each enzyme, the efficiency of synthesis and the potential for peptide synthesis will greatly differ from one protease to another. Strategies of enzymatic synthesis of peptides Despite their good catalytic properties, proteases are not ideal catalysts for the synthesis of peptides. Its specificity and selectivity might limit their potential, particularly in the case of rather large peptides where unwanted hydrolytic reactions will occur over the formed product and the substrates. Besides, the use of non-conventional reaction media and the conditions of temperature and pH required for synthesis can be detrimental both for protease activity and stability (Barberis et al. 2002; Bordusa, 2002; Quiroga et al. 2005; Quiroga et al. 2006). However, there are different strategies to overcome such problems, which comprise the engineering of the reaction medium, the biocatalyst and the substrate (Lombard et al. 2005). Medium engineering: homogeneous, macroheterogeneous and microheterogeneous systems. Medium engineering refers to the rational manipulation of the reaction medium to positively influence the properties of the enzyme with respect to the reaction of synthesis (Clapés et al. 1990a; Clapés et al. 1990b; Khmelnitsky et al. 1991; Ryu and Dordick, 1992; Wescott and Klibanov, 1994; Carrea et al. 1995, Chaudhary et al. 1996). This frequently implies the substitution of the usual aqueous medium for a non conventional medium in which water has been replaced partially or almost totally by another solvent (Hari Krishna, 2002). There are basically two types of biocatalytic systems in non conventional medium: homogeneous systems which are mixtures of water and a water miscible solvent (Castro and Knubovets, 2003) and heterogeneous systems (Krieger et al. 2004) in which a second phase is produced by the presence of a water-immiscible solvent. Heterogeneous systems can be divided into macroheterogeneous, in which two immiscible liquid phases are apparent, and microheterogeneous in which one of the phases (usually the aqueous phase that surrounds the enzyme) is not visible to the naked eye. Both homogeneous and heterogeneous systems can perform with the enzyme dissolved in the medium or insolubilized in it, be it because the enzyme protein is itself insoluble in that medium or because it is immobilized into a solid carrier (Illanes and Barberis, 1994). In this latter case, the system will be always heterogeneous in nature. The replacement of water for another solvent can be beneficial for the enzymatic synthesis of peptides for one or more of several reasons, which apply differently to each of the systems mentioned above, namely the increased solubility of hydrophobic amino acid or peptide substrates, the attenuation of the reverse or competing hydrolytic reactions, the reduction of product hydrolysis, the simplicity of product and/or biocatalyst recovery and the increased biocatalyst thermal stability. Homogeneous systems. Homogeneous systems are composed by a mixture of water and a water-miscible solvent (cosolvent) in which the enzyme is dissolved (Torres and Castro, 2004). Cosolvents are usually detrimental for enzyme activity at moderately high concentrations, because they tend to penetrate the aqueous microenvironment that surrounds the enzyme molecules, altering the pattern of interaction of the enzyme with the solvent and distorting its three-dimensional structure (Klibanov, 1986; Laane et al. 1987a; Laane et al. 1987b; Deetz and Rozzell, 1988; Zaks and Klibanov, 1988a; Zaks and Klibanov, 1988b; Mozhaev et al. 1989; Levitsky et al. 1999). However, polyols and glymes are notable exceptions among cosolvents (Castro, 2000; Illanes and Fajardo, 2001). Another way of circumventing the problem of enzyme deactivation is immobilization, so that it is usual that enzymes are used in immobilized form when performing in homogeneous liquid medium. There are several examples of proteases and other peptide bond forming enzymes which have been successfully employed in peptide synthesis in such media (Guisán et al. 1987; Rodger et al. 1987; Lobell and Schneider, 1998; Ye et al. 1998; Castro, 1999; Illanes et al. 2004). Several studies have reported the effect of cosolvents on the activity and stability of proteases and other enzymes, comparing them with the behaviour in aqueous media (Björup et al. 1996; Lozano et al. 1993; Tyagi and Gupta, 1998; Partridge et al. 1999; Rodakiewicz-Nowak et al. 2000; Ghorbel et al. 2003). In general, both the activity and stability of enzymes is impaired with the exceptions noted above. Heterogeneous systems. Macrohetrogeneous systems are composed by two immiscible liquids. They are termed biphasic systems and are usually composed by water and a hydrophobic solvent (Martinek and Semenov, 1981; Martinek et al. 1981; Kuhl and Jakubke, 1990). Substrates can be dissolved in the organic or in the aqueous phase but, independent of substrates partition, biocatalysis will always occur in the aqueous phase where the protease is. The peptide product formed can be partitioned to the organic phase, which is highly desirable to avoid unwanted hydrolysis and drive the equilibrium toward synthesis (Bordusa, 2002). In some cases the organic phase can be one of the substrates itself (Harbron et al. 1986). Biphasic systems have been extensively used for enzymatic peptide synthesis and represents a good strategy because it is highly flexible and can accommodate to the properties of substrates and products (Kimura et al. 1990a; Kuhl and Jakubke, 1990; Bemquerer et al. 1991; Feliú et al. 1995; Wubbolts et al. 1996; Capellas et al. 1997; Sarmento et al. 1998; Murakami et al. 2000; Barberis et al. 2002; Trusek-Holownia, 2003; Barberis et al. 2006). The main drawback of biphasic systems is the presence of an interphase that can impose diffusional restrictions to the substrates and so reduce the reaction rate. This can be alleviated by intense agitation, but this will in turn promote enzyme inactivation at the interphase (Feliú et al. 1995; Barros et al. 1998). We have not observed this phenomenon when conducting peptide synthesis with phytoproteases (Barberis et al. 2002; Quiroga et al. 2005). Suspension of nearly anhydrous enzymes in hydrophobic solvents can be considered as a microheterogeneous system in the sense that the liquid phase appears as homogeneous to the naked eye; however, the system is microscopically heterogeneous since the solid enzyme is covered by a water shell tightly bound to the enzyme and a bulk hydrophobic organic solvent surrounding it. The enzyme is usually protected from the aggressive hydrophobic solvent by a layer of denatured enzyme (Kanerva and Klibanov, 1989; Klibanov, 1989; Castro, 1999). This is maybe the most simple and most promising strategy for enzymatic synthesis since it exploits to the highest extent the advantages of working in non-conventional medium (Clark, 2004; Gupta and Roy, 2004), namely high thermal stability (Zaks and Klibanov, 1984; Rodger et al. 1987; Klibanov, 2001), potentially favourable changes in substrate specificity (Zaks and Klibanov, 1985; Wescott and Klibanov, 1994; Ebert et al. 1996; Kawashiro et al. 1997; Carrea and Riva, 2000) and easiness of biocatalyst and product recovery (Dickinson and Fletcher, 1989). There are numerous examples of proteases and other enzymes (ribonuclease, pancreatic lipase, terpene cyclase and cytochrome oxidase) that have been proven much more stable in hydrophobic organic solvents than in water (Ayala et al. 1986; Wheeler and Croteau, 1986; Volkin et al. 1991). The enzyme biocatalyst is simply an acetone precipitated enzyme powder or a lyophilized preparation that is suspended in the organic medium in which is completely insoluble (Russell and Klibanov, 1988; Zaks and Klibanov, 1988a). Therefore, immobilization is not necessarily required since the enzyme is already insolubilized in the reaction medium. However, in some cases immobilization to a solid surface can be convenient by offering an increased surface of contact with the substrate and providing additional stabilization. Best results are obtained with highly hydrophobic solvents (log P > 4; where P is the partition coefficient between n-octanol and water) since the intrusion of solvent in the water shell is hindered and so the enzyme is better protected from the direct contact with the organic solvent molecules (Zaks and Klibanov, 1985). The activity of an enzyme in such media is highly dependent from the pH of the aqueous solution from which it was obtained (by precipitation or lyophilization). This has been called pH memory and it stems from the fact that the enzyme retains in the hydrophobic medium the protonation stage of the aqueous solution at which it was exposed previously. This has been elucidated by FTIR spectroscopy (Constantino et al. 1997). Despite its advantages, this strategy has important drawbacks, being the most important the dramatic decrease in activity that enzymes usually express in this kind of media (Klibanov, 1997; Quiroga et al. 2006). However, some improvements in activity have been obtained by the addition of water mimics, such as formamide or ethylene glycol, to the reaction medium or the addition of crown ethers during the preparation of the lyophilized enzyme (van Unen et al. 2001). A 425 fold increase in enzyme activity was observed by the addition of 18-crown-6 to chymotrypsin in the synthesis of a dipeptide in acetonitrile medium (van Unen et al. 1998). In addition, most hydrophobic solvents do not solubilize some of the substrates used in the reactions of synthesis, which also holds for peptide synthesis. Another system that can be considered as microheterogeneous is reverse micelles, which are spontaneously formed when small amounts of water are added to a hydrophobic solvent in the presence of a surfactant and under agitation (Martinek et al. 1986; Gómez-Puyou and Gómez-Puyou, 1998). It has been claimed that the microenvironment in the internal cavity of the micelles is more natural to the enzyme than the bulk aqueous medium, which leads in some cases to an enhanced expression of its catalytic potential (Castro and Cabral, 1989). Reverse micelles have, however, several drawbacks: they are mechanically week, there are no rational methods for its optimization and the surfactant impairs the recovery and purification of products (Bordusa, 2002). t-Boc-Arg-Gly-OEt and Ac-Gly-Asp-diOMe have been successfully synthesized under kinetic control in organic medium using proteases immobilized in reverse micelles (Chen et al. 1998; Chen et al. 1999). An alternative to the classic biphasic and micellar systems has been proposed by Clapés et al. (2001) as reaction medium for peptide synthesis with chymotrypsin using a water-in-oil (W/O) emulsion with a high water content (95%) which is the opposite as in reverse micelles. Such systems have been occasionally used in organic synthesis (Manabe et al. 2000), but its application in biocatalysis is still in its early development. Biocatalyst engineering. Biocatalyst engineering refers to all strategies aimed to obtain biocatalysts well suited to perform under the conditions of synthesis and include approaches that range from chemical modification to genetic and protein engineering (Roig and Kennedy, 1992; Bordusa, 2002; Adamczak and Hari-Krishna, 2004; Hudson et al. 2005). The solubilization of enzymes in nearly anhydrous organic solvents can be attained by their chemical modification. Such modifications may range from simple non-specific acetylation of free amino groups of the enzyme (Murphy and Fagain, 1997) to specific modifications with hydrophilic polymers (Inada et al. 1995; De Santis and Jones, 1999; Salleh et al. 2002) that bind to amphipatic groups at the enzyme surface. The insolubilization of the biocatalyst by immobilization to a solid carrier or by protein aggregation represents the most relevant biocatalyst engineering strategy for producing robust enzyme catalysts well suited to withstand the harsh conditions prevailing during the reactions of synthesis (Adlercreutz, 1991; Wong and Whitesides, 1994; Yan et al. 2002; García-Junceda et al. 2004). Immobilization has been extensively used to produce protease biocatalysts for peptide synthesis (Guisán et al. 1991; Guisán et al. 1997; Filippova and Lysogorskaia, 2003; Lei et al. 2004). Insolubilization by protein cross-linking is a promising technology to produce biocatalysts for synthesis. Cross linked enzyme crystals (CLEC) are robust biocatalysts produced by crystallization of the enzyme protein followed by cross-linking with the bifunctional reagent glutaraldehyde (St. Clair and Navia, 1992; Lalonde, 1997; Abraham et al. 2004). Its specific activity is very high since there is no inert matrix and the whole mass of biocatalyst is essentially pure enzyme protein; however, it has the serious disadvantage of requiring the enzyme in a pure state to be able to crystallize it, which in practice means that the cost of the biocatalyst is very high. CLECs of subtilisin have been recently used successfully in organic synthesis both in repeated batch and continuous operation (Amorim Fernandes et al. 2005). A most promising type of new generation biocatalysts is cross-linked enzyme aggregates (CLEAs) in which the protein solution containing the enzyme is subjected to non-denaturing precipitation by conventional procedures (i.e. salting-out, solvent or polymer precipitation) and then cross-linked with bifunctional reagents (López-Serrano et al. 2002; Shah et al. 2006). CLEAs are similar to CLECs in their properties, with the additional advantage of its simplicity and low cost since the enzyme does not require to be purified to any extent (Cao et al. 2000; Cao et al. 2003). CLEAs prepared by co-aggregation with polyionic polymers (Wilson et al. 2004a) and CLEAs encapsulated into hydrophilic polyvynil alcohol hydrogel (Wilson et al. 2004b) have provided an adequate microenvironment for enzymatic peptide-bond formation in organic synthesis in non-aqueous environments. Protease CLEAs are promising for peptide synthesis even though autoproteolysis can play a role in this case because of the close proximity and flexibility of the enzyme molecules. Other strategies for improving the catalytic performance of enzymes consider the manipulation of the genes encoding the enzyme protein (Arnold, 1990; Jaeger and Reetz, 2000; Reetz and Jaeger, 2000; Arnold, 2001). One relevant strategy is site-directed mutagenesis (sometimes termed as rationale design) which is a powerful tool to analyze structure-activity relationships and also to screen for mutant enzymes with improved properties of activity, stability or specificity. Success of rational design depends on the progress made in structure determination, improved computer modeling protocols and significant new insights into structure-activity relationships (Adamczak and Hari Krishna, 2004). Proteases have been preferred models for applying this technique. In fact, subtilisin has been extensively studied, and considerable progress has been made in engineering this protease and its substrates for peptide bond formation in aqueous solution (Abrahmsen et al. 1991). In P. aeruginosa, a disulfide bond between Cys-30 and Cys-58 played an important role in the organic solvent stability of the PST-01 protease (Ogino et al. 2001). The effect of a novel disulfide bond engineered in subtilisin E from Bacillus sp. based on the structure of a thermophilic subtilisin-type serine protease (aqualysin I) was examined and suggested that an electrostatic interaction between Lys 170 and Gly 195 is important for catalysis and stability in that protease (Takagi et al. 2000). Substitution of native amino acids by fluoroalkyl analogs in the commercially available proteases trypsin and α-chymotrypsin represents a new approach for the design of biologically active peptides with increased stability as well as defined secondary structure, which provides a powerful tool for spectroscopic investigations (Thust and Koksch, 2003). Site-directed mutagenesis has also been employed to improve the properties of trypsin for performing peptide synthesis (Lombard et al. 2005). Other strategy for improving enzyme performance based on genetic manipulation is directed evolution (Arnold, 2001), which is particularly suitable when not enough information on enzyme structure is available. It consists basically in sequential rounds of random mutagenesis using error-prone PCR. At each round, the mutant having the best feature is screened and subjected to the next round. At the end, as a result of this evolutionary progress, the biocatalyst will be improved with respect to the property used for its screening (Gupta and Roy, 2004). Random mutagenesis has been successfully applied to improve the thermal stability of subtilisin (Adamczak and Hari Krishna, 2004). Substrate engineering. Even though most proteases are able to recognize more than one amino acid, not all are able to couple any aminoacidic sequence. Due to the specificity of proteases to a particular amino acid, only those acyl donors that have a specific amino acid in the C-terminal position can be coupled without side reactions. For instance, trypsin requires arginine or lysine residues as carboxylic terminal components in the structure of the acyl donor (Bordusa, 2002). In this way, the manipulation of the leaving group is generally useful to increase the specificity of the protease to a previously less specific amino acid, so increasing reaction rate (Miyazawa et al. 2001a; Miyazawa et al. 2001b). The manipulation of the leaving group affects the aminolysis/hydrolysis ratio of an acyl donor and therefore the conversion yield, since the acyl-enzyme intermediate formed is the same regardless of the change produced in the leaving group. Another approach is the use of mimetic substrates. Contrary to the classic manipulation of the leaving group, focused on the increase in enzyme specificity, mimetic substrates are designed to bind to the active site of the enzyme. In this way, serine and cysteine proteases can react with non specific amino acids or peptide sequences without altering the enzyme or the reaction medium (Schellenberger et al. 1991; Thormann et al. 1999). The main advantage of this strategy is that mimetic substrates allow the formation of the acyl-enzyme and the nucleophile reaction to establish a peptide bond that cannot be further hydrolyzed because it does not correspond to the protease specificity (Bordusa et al. 1997; Lombard et al. 2005). However, this approach is limited to reactions with nonspecific amino acid containing peptides, whereas the coupling of specific ones leads to unwanted cleavages due to the native proteolytic activity of the biocatalyst (Grunberg et al. 2000). Advances and challenges in enzymatic synthesis of peptides The application of proteases to peptide synthesis, although offering several advantages, has been discouraged to some extent by some unfavourable factors. However, several technologies are being actively explored to overcome these difficulties. Low activity of enzymes in non-aqueous solvents, as compared to that in water, has been largely overcome by crown ether treatment of enzymes, crown ether-enhanced enzyme activity being retained even after removal of the crown by washing with a dry organic solvent (Van Unen et al. 2002). Subtilisin Carlsberg in the form of an enzyme-surfactant complex, displayed high catalytic activity in anhydrous hydrophilic organic solvents and did not require excess amount of water, so that the side hydrolytic reaction was effectively suppressed and the yield of the dipeptide formed became considerably high (Okazaki et al. 2000). However, peptide synthesis using organic solvents as reaction medium remains limited to the synthesis of model di-and tripeptides and little effort has been done on the synthesis of bioactive oligopeptides as compared to conventional aqueous systems (Jakubke et al. 1985; Morihara, 1987; Isono and Nakajima, 2002). A general approach to the enzymatic peptide synthesis remains to be formulated despite the large number of studies carried out on the synthesis of model and bioactive peptides. Use of protected substrates in oligopeptide synthesis has led to a loss of continuity in the reaction sequence and a reduction in yield and productivity, limiting the usefulness of enzymes for such purpose. An interesting approach was designed for the enzymatic synthesis of the cholescystokinin C-terminal octapeptide (CCK-8), which is a biologically active peptide of therapeutic value as an agent for the control of gastrointestinal function (Capellas et al. 1996a). The synthesis was performed in organic media and designed in a convergent mode in which a tripeptide and a pentapeptide were synthesized separately using papain, α-chymotrypsin and thermolysin, all commercially available inexpensive proteases (Capellas et al. 1996b; Capellas et al. 1997; Romero et al. 1997; Xiang et al. 2004), and finally the two fragments were linked. The process was scaled up to pilot plant and the economics of the process was assessed (Romero et al. 1997; Ruiz et al. 1997; Fité et al. 2002). The global yield of the process was rather low and the requirement of protection at different stages of the process was a major obstacle to go into full scale operation. However, the strategy may prove useful for other high-value biologically active peptides. Despite the technological advances in peptide synthesis by biocatalysis, low productivity, low yield and high cost of enzymes are problems to be solved to make it competitive in a broad spectrum of cases. More specific, active and stable enzymes are intensively being pursued and the trend should be to improve existing proteases by the use of the modern technologies of genetic and protein engineering and automated screening of proteases from novel microbial strains, mainly extremophiles. Since the pioneering works of du Vigneaud and Merrifield in solid phase synthesis (du Vigneaud et al. 1953; Merrifield, 1963; Merrifield, 1996), the chemical synthesis of peptides has been developed and automated to a high degree in the last decades. Protocols have been well established, which are amenable for scale-up to match the production levels required by the market. A key advantage of the chemical synthesis in solid phase is that the peptide product can be easily separated from impurities and side products. Major drawbacks refer to the racemization during peptide bond formation, the requirement of protection of the side chains of the amino acids that increases the cost of the substrates and reduces the yield of product recovery during deprotection, the difficulty of recycling the coupling reagent and the acyl donor used in excess to achieve rapid and complete acylation of the nucleophile, the time consumed in protection and deprotection reactions that reduce the productivity of the process and the toxic nature of solvents and coupling reagents that may lead to health and environmental concerns (Gill et al. 1996; Nilsson et al. 2005). Despite these restrictions, chemical synthesis of peptides can be considered the most mature technology available, being especially suited for medium size peptides up to one hundred amino acid residues, which comprises most of the peptides of therapeutic relevance. The development of new methods suitable for the large-scale production of biologically active peptides with proteases has been actively pursued over the last decade. However, enzymes are in general labile catalysts, so that process engineering of enzymatic reactions should be designed carefully. This implies the optimization of most relevant operational parameters: pH, temperature, organic solvent concentration, and the assessment of the activity and stability of the biocatalyst under operation conditions, the solubility of reactants, the stability of reactants and products and the selection of conversion yield or productivity as an adequate objective function, or an economic objective functions comprising both (Blanco et al. 1991; Illanes and Wilson, 2003). In this sense, enzymatic synthesis of peptides is a less mature technology than chemical synthesis and no general protocols of synthesis are available, being each situation a particular case that has to be extensively studied and optimized to be technologically competitive. The higher number of critical variables in an enzymatic process makes its optimization cumbersome. However, the limitations of chemical synthesis are generally overcome by protease synthesis. The high specificity and high reactivity under mild operation conditions, which is characteristic of enzymatic processes, can have a strong impact on process economics, since it will reduce the number of operations required for the synthesis, will have less stringent requirement for equipment, will reduce the energy input required and will produce a significantly milder environmental impact as a consequence of the lesser amounts and lower toxicity indexes of the wastes produced. This latter aspect is quite relevant since enzymatic processes can be considered as a clean technology, more in accordance with the concept of sustainable growth. Increasing health and safety regulations and the growing demand for biologically active peptides have prompted an intensive search for biotechnological alternatives to chemical synthesis of peptides of relevance in the medical and food areas. The size of the peptide is a major constraint for the enzymatic synthesis since no established and automated protocols have been yet developed and as the peptide chain growths, requirements of protection and modification of several operational variables at each step make the process cumbersome, so that in practice only small peptides of less than 10 residues have been synthesized enzymatically with moderate success. Of course, the strategy of convergence used in chemical synthesis is also an option for the enzymatic synthesis of peptides, as elegantly shown by the protease synthesis of the CCK octapeptide (Fité et al. 2002). Some relevant examples of enzymatically synthesized biologically active peptides, such as antibiotic and antiviral agents, neuroactive peptides, enzyme regulators and inhibitors, hormones and immunoactive peptides, and functional peptides (sensory, antioxidant, surface active and nutritive peptides) have been highlighted (Gill et al. 1996). The great potential of the enzymatic synthesis of peptides is a powerful driving force for research in the design of the enzyme biocatalysts as well as in the substrate and the reaction medium. Advances in those fields are impressive so that technological outcomes will certainly occur in the forthcoming decades. As for now, competition with the more established technologies of chemical synthesis will be hard, except in particular niches where the outstanding properties of enzymes have profound process implications. This is illustrated by the case of the non-caloric sweetener aspartame, which is increasingly being produced by an enzymatic process with the protease thermolysin (Murakami et al. 2000; Schmid et al. 2001). It is important to point out that the use of organic solvents usually required for performing enzymatic peptide synthesis imposes additional restrictions on process design and engineering (Gill et al. 1996) and contradicts the clean technology concept, so major efforts are being directed toward the replacement by greener systems, among which ionic liquids (Park and Kazlauskas, 2003; Van Rantwijk et al. 2003; Liu et al. 2005) and solid or semisolid reaction media are prominent (Hudson et al. 2005). However, these systems are still in a stage of technological development. Chemical synthesis, especially in solid-phase, can be considered now as the most mature technology for peptide production, being especially suited for medium size peptides among which are the most relevant pharmaceutical and health-care products. Lack of specificity and environmental burden are, however, severe drawbacks of chemical processes that can be successfully overcame by enzyme biocatalysis (Sergeeva et al. 1997). However, strategies for enzymatic synthesis are still in developmental stage and no protocols exist for its validation and scale-up, so that each particular product has to be developed as a case study in which a high number of variables have to be optimized in terms of cost-based objective functions, which is by no means an easy task. As to now, productivity of enzymatic synthesis is not comparable to that of chemical synthesis and in general costs of biocatalysts are still high (Capellas et al. 1996b; Fité et al. 2002). However, the potential of enzymatic processes for peptide synthesis is apparent since most of the present restrictions can be and are being solved through research and development in different aspects of enzyme biocatalysis, while its advantages are there to stay. It is foreseeable then, that enzymatic synthesis of peptides will gain a place of relevance in the forthcoming decades. The combination of chemical and enzymatic synthesis is probably the way to go as already suggested (Hou et al. 2005), since the good properties of each technology can be synergistically used in the context of one process objective. The increasing impact and adoption of biocatalysis in the pharmaceutical and fine-chemical industries will certainly pave the way for this fruitful encounter. This work was done within the framework of Project CYTED IV.22 Industrial Application of Proteolytic Enzymes from Higher Plants. The authors wish to express their thanks to Dr. Nestor Caffini for his encouragement and support and to Ms. Zuly Rivera for her technical assistance in the preparation of the manuscript. ABRAHAM, Tholath Emilia; JOSEPH, Jegan Roy; BINDHU, Laxmi Bai Vasanthakumari and JAYAKUMAR, Kizakoottu Kunjunny. Crosslinked enzyme crystals of glucoamylase as a potent catalyst for biotransformation. Carbohydrate Research, April 2004, vol. 339, no. 6, p. 1099-1104. [CrossRef] ABRAHMSEN, Lars; TOM, Jeffrey; BURNIER, John; BUTCHER, Karen A.; KOSSIAKOFF, Anthony and WELLS, James A. Engineering subtilisin and its substrates for efficient ligation of peptide bond in aqueous solution. Biochemistry, April 1991, vol. 30, no. 17, p. 4151-4159. [CrossRef] ADAMCZAK,
Marek and HARI ALBERICIO, Fernando. Developments in peptide and amide synthesis. Current Opinion in Chemical Biology, June 2004, vol. 8, no. 3, p. 211-221. [CrossRef] ALBERICIO, Fernando; BURGER, Klaus; RUIZ-RODRIGUEZ, Javier and SPENGLER, Jan. A new strategy for solid-phase depsipeptide synthesis using recoverable building blocks. Organic Letters, February 2005, vol. 7, no. 4, p. 597-600. [CrossRef] AMADOR, Roberto; MORENO, Alberto; VALERO, Victoria; MURILLO, Luis; MORA, Ana Lucía; ROJAS, Mauricio; ROCHA, Claudia; SALCEDO, Margarita; GUZMÁN, Fanny; ESPEJO, Fabiola; NUÑEZ, Francisco and PATARROYO, Manuel E. The first field trials of the chemically synthesized malaria vaccine SPf66: safety, immunogenicity and protectivity. Vaccine, 1992, vol. 10, no. 3, p. 179-184. [CrossRef] AMBULOS, Nicholas P.; BIBBS, Lisa; BONEWALD, Lynda F.; KATES, Steven A.; KHATRI, Ashok; MEDZIHRADSZKY, Katalin F. and WEINTRAUB, Susan T. Analysis of Synthetic Peptides. In: KATES, Steven A. and ALBERICIO, Fernando eds. Solid-Phase Synthesis: A Practical Guide. New York, Marcel Dekker Inc., 2000, p. 751-789. AMORIM FERNANDES, João F.; MCALPINE, Morven and HALLING, Peter J. Operational stability of subtilisin CLECs in organic solvents in repeated batch and in continuous operation. Biochemical Engineering Journal, May 2005, vol. 24, no. 1, p. 11-15. [CrossRef] ANDERSSON, Lars; BLOMBERG, Lennart; FLEGEL, Martin; LEPSA, Ludek; NILSSON, Bo and VERLANDER, Michael. Large-scale synthesis of peptides. Biopolymers - Peptide Science Section, 2000, vol. 55, no. 3, p. 227-250. [CrossRef] ARRIBÉRE, María Cecilia; CORTADI, Adriana A.; GATTUSO, Martha A.; BETTIOL, Marisa P.; PRIOLO, Nora S. and CAFFINI, Néstor O. Comparison of Asclepiadaceae latex proteases and characterization of Morrenia brachystephana Griseb. cysteine peptidases. Phytochemical Analysis, November-December 1998, vol. 9, no. 6, p. 267-273. [CrossRef] ARRIBÉRE, María Cecilia; VAIRO CAVALLI, Sandra E.; PRIOLO, Nora S.; CAFFINI, Néstor O.; GATTUSO, Martha A. and CORTADI, Adriana A. Proteolytic enzymes from the latex of Morrenia odorata (Hook et Arn.) Lindley (Asclepiadaceae). Acta Horticulturae, 1999, vol. 501, p. 259-268. ASO, K. Enzymatic approach to the synthesis of a lysine-containing sweet peptide, N-acetyl-Lphenylalanyl-L-lysine. Agricultural and Biological Chemistry, 1989, vol. 53, no. 3, p. 729-733. BACA,
Manuel; MUIR, Tom W.; SCHNÖLZER, Martina and BANFI, Damiano; MUTTER, Manfred and PATINY, Luc. Versatile synthesis of Boc protected hydrazinoacetic acid and its application to the chemoselective ligation of TASP molecules. Protein and Peptide Letters, December 2004, vol. 11, no. 6, p. 539-542. [CrossRef] BARBERIS, Sonia; QUIROGA, Evelina; ARRIBÉRE, Maria Cecilia and PRIOLO, Nora. Peptide synthesis in aqueous-organic biphasic systems catalyzed by a protease isolated from Morrenia brachystephana (Asclepiadaceae). Journal of Molecular Catalysis B: Enzymatic, March 2002, vol. 17, no. 1, p. 39-47. [CrossRef] BARBERIS, Sonia; QUIROGA, Evelina; MORCELLE, Susana; PRIOLO, Nora and LUCO, Juan M. Study of phytoproteases stability in aqueous-organic biphasic systems using linear free energy relationships. Journal of Molecular Catalysis B: Enzymatic, February 2006, vol. 38, no. 2, p. 95-103. [CrossRef] BARLOS, Kleomenis and GATOS, Dimitrios. 9-Fluorenylmethyloxycarbonyl/tbutyl-based convergent protein synthesis. Biopolymers - Peptide Science Section, 1999, vol. 51, no. 4, p. 266-278. [CrossRef] BARRET,
Alan J.; RAWLINGS, Neil D. and WOESSNER, J. Fred. Handbook of Proteolytic
Enzymes. BARROS,
Raúl .J.; WEHTJE, Ernst and ADLERCREUTZ, Patrick. Mass transfer studies
on immobilized α-chymotrypsin biocatalysts prepared by deposition
for use in organic medium. BECKER, Christian F.W.; HUNTER, Christie L.; SEIDEL, Ralf; KENT, Stephen B.H.; GOODY, Roger S. and ENGELHARD, Martin. Total chemical synthesis of a functional interacting protein pair: the protooncogene H-Ras and the Ras-binding domain of its effector c-Raf1. Proceedings of the National Academy of Sciences of the United States of America, April 2003, vol. 100, no. 9, p. 5075-5080. [CrossRef] BEMQUERER, M.P.; THEOBALDO, F.C. and TOMINAGA, M. Pepsin-catalyzed peptide synthesis in biphasic systems. Biomedica Biochimica Acta, 1991, vol. 50, no. 10-11, p. 94-97. BERMUDEZ, A.; CIFUENTES, G.; GUZMAN, F.; SALAZAR, L.M. and PATARROYO, M.E. Immunogenicity and protectivity of Plasmodium falciparum EBA-175 peptide and its analog is associated with alpha-helical region shortening and displacement. Biological Chemistry, October-November 2003, vol. 384, no. 10-11, p: 1443-1450. BJÖRUP, Peter; WEHTJE, Ernst and ADLERCREUTZ, Patrick. Effects of acetonitrile-water mixtures on α-chymotrypsin catalysed dipeptide synthesis. Biocatalysis and Biotransformation, 1996, vol. 13, no. 3, p. 189-200. BJÖRUP, Peter; ADLERCREUTZ, Patrick and CLAPÉS, P. Useful methods in enzymatic synthesis of peptides: a comparative study focusing on kinetically controlled synthesis of Ac-Phe-Leu-NH2 catalyzed by α-chymotrypsin. Biocatalysis and Biotransformation, 1999, vol. 17, no. 5, p. 319-345. BLANCO, Rosa M.; ALVARO, Gregorio and GUISÁN, José M. Enzyme reaction engineering: design of peptide synthesis by stabilized trypsin. Enzyme and Microbial Technology, July 1991, vol. 13, no. 7, p. 573-583. [CrossRef] BORDUSA, Frank; ULLMANN, Dirk; ELSNER, Christian and JAKUBKE, Hans-Dieter. Substrate mimetics mediated peptide synthesis: an irreversible ligation strategy that is independent of substrate specificity. Angewandte Chemie - International Edition in English, December 1997, vol. 36, no. 22, p. 2473-2475. [CrossRef] BORDUSA, Frank. Proteases in organic synthesis. Chemical Reviews, December 2002, vol. 102, no. 12, p. 4817-4868. [CrossRef] BORNSCHEUER, Uwe Theo and KAZLAUSKAS, Romas Joseph. Hydrolases in Organic Synthesis: Regio- and Stereoselective Biotransformation. Weinheim, Wiley-VCH, 1999. 336 p. ISBN 3-527-30104-6. BRAHMS, S. and BRAHMS, J. Determination of protein secondary structure in solution by vacuum ultraviolet circular dichroism. Journal of Molecular Biology, April 1980 vol. 138, no. 2, p. 149-178. [CrossRef] BRAY, Brian L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nature Reviews Drug Discovery, July 2003, vol. 2, no. 7, p. 587-593. [CrossRef] BROOS, Jaap; ENGBERSEN, Johan F.J.; SAKODINSKAYA, Inna K.; VERBOOM, Willem and REINHOUDT, David N. Activity and enantioselectivity of serine proteases in transesterification reactions in organic media. Journal of Chemical Society Perkin Transactions 1, 1995, vol. 1. p. 2899-2905. BRUCKDORFER, Thomas; MARDER, Oleg and ALBERICIO, Fernando. From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future. Current Pharmaceutical Biotechnology, February 2004, vol. 5, no. 1, p. 29-43. CALVO, J.C.; RIVERA, Z.J.; CARVAJAL, C.C.; BERNAL, A.; CHAVES, F.A.; LOZANO, J.M.; TRUJILLO, M.; GUZMAN, F. and PATARROYO, M.E. Síntesis de péptidos dendriméricos usando la estrategia de doble dímero. Revista Colombiana de Química, 1999, vol. 28, no. 1, p. 27-34. CALVO, Julio C.; CHOCONTA, Katia C.; DIAZ, Diana; OROZCO, Oscar; BRAVO, Maria M.; ESPEJO, Fabiola; SALAZAR, Luz; GUZMAN, Fanny and PATARROYO, Manuel E. An alpha helix conformationally restricted peptide is recognized by cervical carcinoma patients' sera. Journal of Medicinal Chemistry, December 2003, vol. 46, no. 25, p. 5389-5394. [CrossRef] CANNE, Lynne E.; BOTTI, Paolo; SIMON, Reyna J.; CHEN, Yijun; DENNIS, Edward A. and KENT, Stephen B. Chemical protein synthesis by solid phase ligation of unprotected peptide segments. Journal of the American Chemical Society, September 1999, vol. 121, no. 38, p. 8720-8727. [CrossRef] CAO, Linqiu; VAN RANTWIJK, Fred and SHELDON, Roger A. Cross-linked enzyme aggregates: a simple and effective method for the immobilization of penicillin acylase. Organic Letters, May 2000, vol. 2, no. 10, p. 1361-1364. [CrossRef] CAO, Linqiu; VAN LANGEN, Luuk and SHELDON, Roger A. Immobilised enzymes: carrier-bound or carrier free? Current Opinion in Biotechnology, August 2003, vol. 14, no. 4, p. 387-394. [CrossRef] CAPELLAS, Montserrat; BENAIGES, M. Dolors; CAMINAL, Gloria; GONZÁLEZ, Gloria; LÓPEZ-SANTÍN, Josep and CLAPÉS, Pere. Influence of water activity and support material on the enzymatic synthesis of a CCK-8 tripeptide fragment. Biocatalysis and Biotransformation, 1996a, vol. 13, no. 3, p. 165-178. CAPELLAS, Montserrat; BENAIGES, M. Dolors; CAMINAL, Gloria; GONZÁLEZ, Gloria; LÓPEZ-SANTÍN, Josep and CLAPÉS, Pere. Enzymatic synthesis of a CCK-8 tripeptide fragment in organic media. Biotechnology and Bioengineering, June 1996b, vol. 50, no. 6, p. 700-708. [CrossRef] CAPELLAS,
CARREA, Giacomo; OTTOLINA, Gianluca and RIVA, Sergio. Role of solvents in the control of enzyme selectivity in organic media. Trends in Biotechnology, February 1995, vol. 13, no. 2, p. 63-70. [CrossRef] CARREA, Giacomo and RIVA, Sergio. Properties and synthetic applications of enzymes in organic solvents. Angewandte Chemie - International Edition, July 2000, vol. 39, no. 13, p. 2226-2254. [CrossRef] CARREA, Giacomo; COLONNA, Stefano; KELLY, David R.; LAZCANO, Antonio; OTTOLINA, Gianluca and ROBERTS, Stanley M. Polyamino acids as synthetic enzymes: mechanism, applications and relevance to prebiotic catalysis. Trends in Biotechnology, October 2005, vol. 23, no. 10, p. 507-513. [CrossRef] CASSELLS, John M. and HALLING, Peter J. Protease-catalysed peptide synthesis in aqueous-organic two-phase systems: reactant precipitation and interfacial inactivation. Enzyme and Microbial Technology, October 1990, vol. 12, no. 10, p. 755-759. [CrossRef] CASTRO, M.J.M. and CABRAL, J.M.S. Kinetic studies of hydrogenase in AOT reversed micelles. Enzyme and Microbial Technology, January 1989, vol. 11, no. 1, p. 6-11. [CrossRef] CASTRO, Guillermo R. Enzymatic activities of proteases dissolved in organic solvents. Enzyme and Microbial Technology, November 1999, vol. 25, no. 8-9, p. 689-694. [CrossRef] CASTRO, Guillermo R. Properties of soluble α-chymotrypsin in neat glycerol and water. Enzyme and Microbial Technology, July 2000, vol. 27, no. 1-2, p. 143-150. [CrossRef] CASTRO, Guillermo R. and KNUBOVETS, Tatyana. Homogeneous biocatalysis in organic solvents and water-organic mixtures. Critical Reviews in Biotechnology, July-September 2003, vol. 23, no. 3, p. 195-231. CHAN,
Wing C. and WHITE, Peter D. Fmoc solid phase peptide synthesis:
a practical approach. CHAUDHARY, Apurva K.; KAMAT, Sanjav V.; BECKMAN, Eric J.; NUROK, David; KLEYLE, Robert M.; HAJDU, Paul and RUSSELL, Alan J. Control of subtilisin substrate specificity by solvent engineering in organic solvents and supercritical fluoroform. Journal of the American Chemical Society, 1996, vol. 118, no. 51, p. 12891-12901. [CrossRef] CHÁVEZ, F.; CALVO, J.C.; CARVAJAL, C.; RIVERA, Z.; RAMÍREZ, L.; PINTO, M.; TRUJILLO, M.; GUZMAN, F. and PATARROYO, M.E. Synthesis, isolation and characterization of Plasmodium falciparum antigenic tetrabranched peptide dendrimers obtained by thiazolidine linkages. Journal of Peptide Research, October 2001, vol. 58, no. 4, p. 307-316. [CrossRef] CHELLAPAN, S.; JASMIN, C.; BASHEER, S.; ELYAS, K.K., BHAT, S.G. and CHANDRASEKARAN, M. Production, purification and partial characterization of a novel protease from marine Engyodontium album BTMFS 10 under solid state fermentation. Process Biochemistry, April 2006, vol. 41, no. 4, p. 956-961. [CrossRef] CHEN, Shui-Tein; CHEN, Shiah-Yun; KAO, Chai-Lin and WANG, Kung-Tsung. Improved yield by cryoeffect in kinetically controlled peptide synthesis catalyzed with alcalase. Biotechnology Letters, October 1994, vol. 16, no. 10, p. 1075-1080. [CrossRef] CHEN, Yu-Xin; ZHANG, Xue-Zhong; ZHENG, Kun; CHEN, Song-Ming; WANG, Qian-Chuan and WU, Xiao-Xia. Protease-catalyzed synthesis of precursor dipeptides of RGD with reverse micelles. Enzyme and Microbial Technology, August 1998, vol. 23, no. 3, p. 243-248. [CrossRef] CHEN, Yu-Xin; ZHANG, Xue-Zhong; CHEN, Song-Ming; YOU, De-Lin; WU, Xiao-Xia; YANG, Xue-Cheng and GUAN, Wen-Zeng. Kinetically controlled synthesis catalyzed by proteases in reverse micelles and separation of precursor dipeptides of RGD. Enzyme and Microbial Technology, August 1999, vol. 25, no. 3, p. 310-315. [CrossRef] CHIVA, Cristina; BARTHE, Philippe; CODINA, Anna; GAIRI, Margarida; MOLINA, Franck; GRANIER, Claude; PUGNIÈRE, Martine; INUI, Tatsuya; NISHIO, Hideki; NISHIUCHI, Yuji; KIMURA, Terutoshi; SAKAKIBARA, Shumpei; ALBERICIO, Fernando and GIRALT, Ernest. Synthesis and NMR structure of p41icf, a potent inhibitor of human cathepsin L. Journal of the American Chemical Society, January 2003, vol. 125, no. 6, p. 1508-1517. [CrossRef] CLAPÉS, Pere; MATA-ALVAREZ, Joan; VALENCIA, Gregorio; REIG, Francesca; TORRES, Josep L. and GARCÍA-ANTÓN, Jose-Maria. Continuous enzymatic synthesis of Z-kyotorphin amide in an enzyme-immobilized fixed-bed reactor. Journal of Chemical Technology and Biotechnology, 1989, vol. 45, no. 3, p. 191-202. CLAPÉS, Pere; ADLERCREUTZ, Patrick and MATTIASSON, Bo. Enzymatic peptide synthesis in organic media: a comparative study of water-miscible and water-immiscible solvent systems. Journal of Biotechnology, September 1990a, vol. 15, no. 4, p. 323-338. [CrossRef] CLAPÉS, Pere; ADLERCREUTZ, Patrick and MATTIASSON, Bo. Enzymatic peptide synthesis in organic media: Nucleophile specificity and medium engineering in α-chymotrypsin-catalyzed reactions. Biotechnology and Applied Biochemistry, 1990b, vol. 12, no. 4, p. 376-386. CLAPÉS, Pere; ESPELT, Laia; NAVARRO, M. Antonia and SOLANS, Conxita. Highly concentrated water-in-oil emulsions as novel reaction media for protease-catalysed kinetically controlled peptide synthesis. Journal of Chemical Society Perkin Transactions 2, 2001, vol. 2, no. 8, p. 1394-1399. [CrossRef] CLARK,
Douglas S. Characteristics of nearly dry enzymes in organic solvents:
implications for biocatalysis in the absence of water. Philosophical
Transactions of the Royal Society CLOUET, Anthony; DARBRE, Tamis and REYMOND, Jean-Louis. A combinatorial approach to catalytic peptides dendrimers. Angewandte Chemie - International Edition in English, September 2004, vol. 43, no. 35, p. 4612-4615. [CrossRef] CONSTANTINO, Henry R.; GRIEBENOW, Kai; LANGER, Robert and KLIBANOV, Alexander M. On the pH memory of lyophylized compounds containing protein functional groups. Biotechnology and Bioengineering, February 1997, vol. 53, no. 3, p. 345-348. [CrossRef] CRUZ,
Luis J.; INSUA, Marta Martinez; PEREZ BAZ, Julia; TRUJILLO, Martha;
RODRIGUEZ-MIAS, Richard A.; OLIVEIRA, Eliandre; GIRALT, Ernest; ALBERICIO,
Fernando and CAÑEDO Librada M. IB- CUNNINGHAM, C. and PORTER, A.J. Methods in Biotechnology, Vol.3: Recombinant proteins from plants production and isolation of clinically useful compounds. Totowa, N.J; Humana Press, 1997. 308 p. ISBN: 0-89-603390-2. DAWSON,
P.E.; MUIR, T.W.; CLARK-LEWIS, I. and DAWSON, Philip E. and KENT, Stephen B.H. Synthesis of native proteins by chemical ligation. Annual Reviews of Biochemistry, 2000, vol. 69, no. 1, p. 923-960. [CrossRef] DE SANTIS, Grace and JONES, J. Bryan. Chemical modification of enzymes for enhanced functionality. Current Opinion in Biotechnology, August 1999, vol.10, no. 4, p. 324-330. [CrossRef] DEETZ, J.A. and ROZZELL, D. Enzyme-catalysed reactions in non-aqueous media. Trends in Biotechnology, January 1988, vol. 6, no. 1, p. 15-19. [CrossRef] DICKINSON, Mark and FLETCHER, Paul D.I. Enzymes in organic solvents. Enzyme and Microbial Technology, January 1989, vol. 11, no. 1, p. 55-56. [CrossRef] DRAUZ, Karlheinz and WALDMANN, Herbert. Enzyme Catalysis in Organic Synthesis: A Comprehensive Handbook. 2nd ed. Weinheim; Wiley-VCH, 2002. 1500 p. ISBN 3-527-29949-1. DU VIGNEAUD, Vincent; RESSLER, Charlotte; SWAN, College John M.; ROBETS, Carleton W.; KATSOYANNIS, Panayotis G. and GORDON, Samuel. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. Journal of the American Chemical Society, October 1953, vol. 75, no. 19, p. 4879-4880. [CrossRef] EBERT, Cynthia; GARDOSSI, Lucia; LINDA, Paolo; VESNAVER, Regina and BOSCO, Marco. Influence of organic solvents on enzyme chemoselectivity and their role in enzyme-substrate interaction. Tetrahedron, March 1996, vol. 52, no. 13, p. 4867-4876. [CrossRef] EHRLICH, Angelika; HEYNE, Hans-Ulrich; WINTER, Rüdiger; BEYERMANN, Michael; HABER, Hanka; CARPINO, Louis A. and BIENERT, Michael. Cyclization of all-L-pentapeptides by means of 1-Hydroxy-7-azabenzotriazole-derived uronium and phosphonium reagents. Organic Chemistry, 1996, vol. 61, no. 25, p. 8831-8838. ELLMAN, George L. A colorimetric method for determining low concentrations of mercaptans. Archives of Biochemistry and Biophysics, April 1958, vol. 74, no. 2, p. 443-450. [CrossRef] ERBELDINGER, Markus; NI, Xiongwei and HALLING, Peter J. Enzymatic synthesis with mainly undissolved substrates at very high concentrations. Enzyme and Microbial Technology, July 1998, vol. 23, no. 1, p. 141-148. [CrossRef] FELIÚ, Josep Anton; DE MAS, Carles and LÓPEZ-SANTÍN, Josep. Studies on papain action in the synthesis of Gly-Phe in two-liquid-phase media. Enzyme and Microbial Technology, October 1995, vol. 17, no. 10, p. 882-887. [CrossRef] FILIPPOVA, I.I. and LYSOGORSKAIA, E.N. Modified proteinases in peptide synthesis in organic media. Bioorganicheskaia Khimiia, September-October 2003, vol. 29, no. 5, p. 544-550. FILLON, Yannick A.; ANDERSON, Jason P. and CHMIELEWSKI, Jean. Cell penetrating agents based on a polyproline helix scaffold. Journal of the American Chemical Society, 2005, vol. 127, no. 33, p. 11798-11803. [CrossRef] FITÉ, M.; ALVARO, G.; CLAPÉS, P.; LÓPEZ-SANTÍN, J.; BENAIGES, M.D. and CAMINAL, G. Reactivity of easily removable protecting groups for glycine in peptide synthesis using papain as catalyst. Enzyme and Microbial Technology, August 1998, vol. 23, no. 3-4, p. 199-203. [CrossRef] FITÉ, Mercé; CLAPÉS, Pere; LÓPEZ-SANTÍN, Josep; BENAIGES, M. Dolors and CAMINAL, Glòria. Integrated process for the enzymatic synthesis of the octapeptide PhAcCCK-8. Biotechnology Progress, 2002, vol. 18, no. 6, p. 1214-1220. [CrossRef] FRANK, Ronald. The SPOT-synthesis technique: Synthetic peptide arrays on membrane supports-principles and applications. Journal of Immunological Methods, September 2002, vol. 267, no.1, p.13-26. [CrossRef] GARCÍA-JUNCEDA, Eduardo; GARCÍA-GARCÍA, Juan Francisco; BASTIDA, Agatha and FERNÁNDEZ-MAYORALAS, Alfonso. Enzymes in the synthesis of bioactive compounds: the prodigious decades. Bioorganic and Medicinal Chemistry, April 2004, vol. 12, no. 8, p. 1817-1834. [CrossRef] GHORBEL, Basma; SELLAMI-KAMOUN, Alya and NASRI, Moncef. Stability studies of protease from Bacillus cereus BG1. Enzyme and Microbial Technology, April 2003, vol. 32, no. 5, p. 513-518. [CrossRef] GILL, Iqbal and VULFSON, Evgeny N. Enzymic catalysis in heterogeneous eutectic mixtures of substrates. Trends in Biotechnology, April 1994, vol. 12, no. 4, p. 118-122. GILL, Iqbal; LÓPEZ-FANDIÑO, Rosina L.; JORBA, Xabier and VULFSON, Evgeny N. Biologically active peptides and enzymatic approaches to their production. Enzyme and Microbial Technology, February 1996, vol. 18, no. 3, p. 162-183. GISIN, B.F. and MERRIFIELD, R.B. Carboxyl- catalyzed intramolecular aminolysis. A side reaction in solid-phase peptide synthesis. Journal of the American Chemical Society, May 1972, vol. 94, no. 9, p. 3102-3106. [CrossRef] GÓMEZ-PUYOU, M.T. and GÓMEZ-PUYOU, A. Enzymes in low water systems. Critical Reviews in Biochemistry and Molecular Biology, February 1998, vol. 33, no. 1, p. 53-89. [CrossRef] GOODMAN,
M. Houben-Weyl Methods of Organic Chemistry. Volume E22a. Synthesis
of peptides and peptidomimetics. GOODMAN, Murray; ZAPF, Christoph and REW, Yosup. New reagents, reactions, and peptidomimetics for drug design. Biopolymers - Peptide Science Section, 2001, vol. 60, no. 3, p. 229-245. [CrossRef] GRUNBERG, Raik; DOMGALL, Ines; GUNTHER, Robert; RALL, Kathrin; HOFMAN, Hans-Jörg and BORDUSA, Frank. Peptide bond formation mediated by substrate mimetics: structure-guided optimization of trypsin for synthesis. European Journal of Biochemistry, December 2000, vol. 267, no. 24, p. 7024-7030. [CrossRef] GUICHARD, G. and SEEBACH, D. Solid-phase synthesis of b-oligopeptides. Chimia, 1997, vol. 51, no. 6, p. 315-318. GUISÁN,
J.M.; ALVARO, G. and BLANCO, R.M. Effect of water-miscible organic
solvents on the catalytic activity of penicillin acylase from Kluyvera
citrophila. In: LAANE, C.; TRAMPER, J. and LILLY, M.D. eds.
Biocatalysis in Organic Media. Elsevier Science Publishing Company,
Inc., GUISÁN, José M.; BASTIDA, Agatha; CUESTA, Carmen; FERNÁNDEZ-LAFUENTE, Roberto and ROSELL, Cristina M. Immobilization-stabilization of α-chymotrypsin by covalent attachment to aldehyde-agarose gels. Biotechnology and Bioengineering, December 1991, vol. 38, no. 10, p. 1144-1152. [CrossRef] GUISÁN, J.M.; POLO, E.; AGUADO, J.; ROMERO, M.; ALVARO, G. and GUERRA, M.J. Immobilization-stabilization of thermolysin onto activated agarose gels. Biocatalysis and Biotransformation, 1997, vol. 15, no. 3, p. 159-173. GUPTA, Munishwar N. and ROY, Ipsita. Enzymes in organic media: forms, functions and applications. European Journal of Biochemistry, July 2004, vol. 271, no. 13, p. 2575-2583. [CrossRef] GUZMÁN, Fanny; JARAMILLO, Katherine; SALAZAR, Luz M.; TORRES, Angela; RIVERA, Augusto and PATARROYO, Manuel E. 1H- NMR structures of the Plasmodium falciparum 1758 erythrocyte binding peptide analogues and protection against malaria. Life Sciences, October 2002, vol. 71, no. 23, p. 2773-2785. [CrossRef] HACKENG, Tilman M.; FERNANDEZ, José A.; DAWSON, Philip E.; KENT, Stephen B.H. and GRIFFIN, John H. Chemical synthesis and spontaneous folding of a multidomain protein: anticoagulant microprotein S. Proceedings of the National Academy of Sciences of the United States of America, December 2000, vol. 97, no. 26, p. 14074-14078. [CrossRef] HALLING, Peter J. Thermodynamic predictions for biocatalysis in nonconventional media: Theory, tests, and recommendations for experimental design and analysis. Enzyme and Microbial Technology, March 1994, vol. 16, no. 3, p. 178-206. [CrossRef] HALLING, P.J.; EICHHORN, U.; KUHL, P. and JAKUBKE, H.-D. Thermodynamics of solid-to-solid conversion and application to enzymic peptide synthesis. Enzyme and Microbial Technology, 1995, vol. 17, no. 7, p. 601-606. [CrossRef] HAMEL, E. and COVELL, D.G. Antimitotic peptides and depsipeptides. Current Medicinal Chemistry - Anti-Cancer Agents, January 2002, vol. 2, no. 1, p. 19-53. [CrossRef] HARBRON, S.; SMITH, B.W. and LILLY, M.D. Two-liquid phase biocatalysis: epoxidation of 1,7-octadiene by Pseudomonas putida. Enzyme and Microbial Technology, February 1986, vol. 8, no. 2, p. 85-88. [CrossRef] HARI-KRISHNA, Sajja. Developments and trends in enzyme catalysis in nonconventional media. Biotechnology Advances, November 2002, vol. 20, no. 3-4, p. 239-267. [CrossRef] HERRMANN, G.; SCHWARZ, A.; WANDREY, C.; KULA, M.R.; KNAUP, G.; DRAUZ, K.H. and BERNDT, H. Scale up of enzymatic peptide synthesis in an enzyme membrane reactor. Biotechnology and Applied Biochemistry, 1991, vol. 13, p. 346-353. HIEBL, J.; ALBERTS, D.P.; BANYARD, A.F.; BARESCH, K.; BAUMGARTNER,H.; BERNWIESER, I.; BHATNAGAR, P.K.; BLANKA, M.; BODENTEICH, M.; CHEN, T.; ESCH, P.M.; KOLLMANN, H.; LANTOS, I.; LEITNE,R K.; MAYRHOFER, G.; PATEL ,R.; RIO A.; ROVENSZKY, F.; STEVENSON, D.; TUBMAN, K.D.; UNDHEIM, K.; WEIHTRAGER, H.; WELZ, W. and WINKLER, K. Large-scale synthesis of hematoregulatory nonapeptide SK&F 107647 by fragment coupling. The Journal of Peptide Research, July 1999, vol. 54, no. 1, p. 54-65. [CrossRef] HIPKISS, A.R. and BROWNSON, C. A possible new role for the anti-ageing peptide carnosine. Cellular and Molecular Life Sciences, May 2000, vol. 57, no. 5, p. 747-753. [CrossRef] HOMANDBERG, G.A.; MATTIS, J.A. and LASKOWSKI, M. Synthesis of peptide bonds by proteinases. Addition of organic cosolvents shifts peptide bond equilibria toward synthesis. Biochemistry, 1978, vol. 17, no. 24, p. 5220-5227. HOU, Rui-Zhen; ZHANG, Na; LI, Gang; HUANG, Yi-Bing; WANG, Hua; XIAO, Yu-Peng; LIU, Yun-Jia; YANG, Yan; ZHAO, Lang and ZHANG, Xue-Zhong. Synthesis of tripeptide RGD amide by a combination of chemical and enzymatic methods. Journal of Molecular Catalysis B: Enzymatic, December 2005, vol. 37, no. 1-6, p. 9-15. [CrossRef] HOUGHTEN, Richard A. General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proceedings of the National Academy of Sciences of the United States of America, August 1985, vol. 82, no. 15, p. 5131-5135. HOUGHTEN, Richard A.; WILSON, Darcy B. and PINILLA, Clemencia. Drug discovery and vaccine development using mixture-based synthetic combinatorial libraries. Drug Discovery Today, July 2000, vol. 5, no. 7, p. 276-285. [CrossRef] HUDSON, Elton P.; EPPLER, Ross K. and CLARK, Douglas S. Biocatalysis in semi-aqueous and nearly anhydrous conditions. Current Opinion in Biotechnology, December 2005, vol. 16, no. 6, p. 637-643. [CrossRef] HULTIN, H.O. Current and potential uses of immobilized enzymes. Food Technology, 1983, vol. 37, no. 10, p. 66-70. ILLANES, A. and BARBERIS, S.E. Catálisis enzimática en fase orgánica. In: Biotecnología de Enzimas. Monografía 35. Serie Biología. Secretaría General de la Organización de los Estados Americanos, Washington, DC, USA. Ediciones Universitarias de Valparaíso, Valparaíso, 1994, p. 225-254. ILLANES, Andrés. Stability of biocatalysts. Electronic Journal of Biotechnology [online]. 15 April 1999, vol. 2, no. 1. Available from: http://www.ejbiotechnology.info/content/vol2/issue1/full/2/index.html. ISSN 0717-3458. ILLANES, Andrés and FAJARDO, Adrián. Kinetically controlled synthesis of ampicillin with immobilized penicillin acylase in the presence of organic cosolvents. Journal of Molecular Catalysis B: Enzymatic, January 2001, vol. 11, no. 4-6, p. 587-595. [CrossRef] ILLANES, Andrés and WILSON, Lorena. Enzyme reactor design under thermal inactivation. Critical Reviews in Biotechnology, January-March 2003, vol. 23, no. 1, p. 61-93. ILLANES, A.; ANJARÍ, S.; ALTAMIRANO, C. and AGUIRRE, C. Optimization of cephalexin synthesis with immobilized penicillin acylase in ethylene glycol medium at low temperatures. Journal of Molecular Catalysis B: Enzymatic, August 2004, vol. 30, no. 2, p. 95-103. [CrossRef] INADA, Yuji; FURUKAWA, Makoto; SASAKI, Hideyuji; KODERA, Yoh; HIROTO, Misao; NISHIMURA, Hiroyuki and MATSUSHIMA, Ayako. Biomedical and biotechnological applications of PEG- and PM-modified proteins. Trends in Biotechnology, March 1995, vol. 13, no. 3, p. 86-91. [CrossRef] ISONO, Yasuyuki and NAKAJIMA, Mitsutoshi. Synthesis of aspartame precursor using proteases suspended in microaqueous molten amino acids mixture. Biocatalysis and Biotransformation, 2002, vol. 20, no. 6, p. 391-395. [CrossRef] JAEGER, Karl-Erich and REETZ, Manfred T. Directed evolution of enantioselective enzymes for organic chemistry. Current Opinion in Chemical Biology, February 2000, vol. 4, no. 1, p. 68-73. [CrossRef] JAKUBKE, Hans-Dieter; KUHL, Peter and KONNECKE, Andreas. Basic principles of protease-catalized peptide bond formation. Angewandte Chemie - International Edition in English, February 1985, vol. 24, no. 2, p. 85-93. [CrossRef] JAKUBKE,
H.D. Enzymatic Peptide Synthesis. In: UDENFRIEND, S. and MEIENHOFER,
J. eds. The Peptides: Analysis, Synthesis, Biology. Academic
Press Inc., JOHNSON, Erik C. and KENT, Stephen B.H. Insights into the mechanism and catalysis of the native chemical ligation reaction. Journal of the American Chemical Society, May 2006, vol. 128, no. 20, p. 6640-6646. [CrossRef] KALIA, A. and GUPTA, R.P. Proteomics: a paradigm shift. Critical Reviews in Biotechnology, October-December 2005, vol. 25, no. 4, p. 173-198. KAMAT, Sanjay; BECKMAN, Eric J. and RUSSELL, Alan J. Role of diffusion in nonaqueous enzymology. 1. Theory. Enzyme and Microbial Technology, April 1992, vol. 14, no. 4, p. 265-271. [CrossRef] KANERVA, L.T. and KLIBANOV, A.M. Hammett analysis of enzyme action in organic solvents. Journal of the American Chemical Society, 1989, vol. 111, p. 6864-6865. KAPLAN, J. and DEGRADO, W.F. De novo design of catalytic peptides. Proceedings of the National Academy of Sciences of the United States of America, August 2004, vol. 101, no. 32, p. 11566-11570. [CrossRef] KASCHE, V. Proteases in peptide synthesis. In: BEYNON, R.J. and NOND, J.S. eds. Proteolytic enzymes a practical approach, IRL Press, Oxford, 1996, p. 125-143. KATZEN, Federico; CHANG, Geoffrey and KUDLICKI, Wieslaw. The past, present and future of cell-free protein synthesis. Trends in Biotechnology, March 2005, vol. 23, no. 3, p. 150-156. [CrossRef] KAWASHIRO, Katsuhiro; SUGAHARA, Hideki; SUGIYAMA, Shigeru and HAYASHI, Horomu. Effect of organic solvents on enantioselectivity of protease catalysis. Biotechnology and Bioengineering, 1997, vol. 53, no. 1, p. 26-31. [CrossRef] KHMELNITSKY, Yuri L.; BUDDE, Cheryl; ARNOLD, Michael; USYATINSKY, Alexander; CLARK, Douglas S. and DORDICK, Jonathan S. Synthesis of water-soluble paclitaxel derivatives by enzymatic acylation. Journal of the American Chemical Society, November 1997, vol. 119, no. 47, p. 11554-11555. [CrossRef] KIMMERLIN, Thierry and SEEBACH, Dieter. 100 years of peptide synthesis': ligation methods for peptide and protein synthesis with applications to b-peptide assemblies. Journal of Peptide Research, February 2005, vol. 65, no. 2, p. 229-260. [CrossRef] KIMMERLIN, Thierry; SEEBACH, Dieter and HILVERT, Donald. Synthesis of β3-peptides and α/β3-peptides by thioligation. Helvetica Chimica Acta, June 2002, vol. 85, no. 6, p. 1812-1826. [CrossRef] KIMURA, Yukitaka; NAKANISHI, Kazuhiro and MATSUNO, Ryuichi. Enzymatic synthesis of the precursor of Leu-enkephalin in water-immiscible organic solvent systems. Enzyme and Microbial Technology, April 1990a, vol. 12, no. 4, p. 272-280. [CrossRef] KIMURA, Yukitaka; YOSHIDA, Tomoko; MURAYA, Koji; NAKANISHI, Kazuhiro and MATSUNO, Ryuichi. Continuous synthesis of a tripeptide by successive condensation and transesterification catalyzed by two immobilized proteinases in organic solvent. Agricultural and Biological Chemistry, June 1990c, vol. 54, no. 6, p. 1433-1440. KIMURA, Yukitaka; MURAYA, Koji; ARAKI, Yoichi; MATSUOKA, Hiroyoshi; NAKANISHI, Kazuhiro and MATSUNO, Ryuichi. Synthesis of peptides consisting of essential amino acids by a reactor system using three proteinases and an organic solvent. Agricultural and Biological Chemistry, December 1990b, vol. 54, no. 12, p. 3331-3333. KIRCHNER, Gerald; SCOLLAR, Mark P. and KLIBANOV, Alexander M. Resolution of racemic mixtures via lipase catalysis in organic solvents. Journal of the American Chemical Society, November 1985, vol. 107, no. 24, p. 7072-7076. [CrossRef] KLEIN, Michael D. and LANGER, Robert. Immobilized enzymes in clinical medicine: an emerging approach to new drug therapies. Trends in Biotechnology, July 1986, vol. 4, no. 7, p. 179-186. [CrossRef] KLIBANOV, Alexander M. Enzymes that work in organic solvents. Chemical Technology, June 1986, vol. 16, no. 6, p. 354-359. KLIBANOV, Alexander M. Why are enzymes less active in organic solvents than in water? Trends in Biotechnology, March 1997, vol. 15, no. 3, p. 97-101. [CrossRef] KLIBANOV, Alexander M. Enzymatic catalysis in anhydrous organic solvents. Trends in Biochemical Sciences, April 1989, vol. 14, no. 4, p. 141-144. [CrossRef] KLIBANOV, Alexander M. Improving enzymes by using them in organic solvents. Nature, January 2001, vol. 409, no. 6817, p. 241-246. [CrossRef] KOFOED, Jacob; DARBRE, Tamis and REYMOND, Jean-Louis. Artificial aldolases from peptide dendrimer combinatorial libraries. Organic and Biomolecular Chemistry, 2006, vol. 4, no. 17, p. 3268-3281. [CrossRef] KRCHÑAK, V.; VAGNER, J. and LEBL, M. Noninvasive continuous monitoring of solid-phase peptide synthesis by acid-base indicator. International Journal of Peptide and Protein Research, November 1988, vol. 32, no. 5, p. 415-416. KRIEGER, Nadia; BHATNAGAR, Tej; BARATTI, Jacques C.; BARON, Alessandra M.; DE LIMA, Valéria M. and MITCHELL, David. Non-aqueous biocatalysis in heterogeneous solvent systems. Food Technology and Biotechnology, October 2004, vol. 42, no. 4, p. 279-286. KUHL, P. and JAKUBKE, H.-D. Protease-catalyzed synthesis of peptides in biphasic aqueous-organic systems. Die Pharmazie, June 1990, vol. 45, no. 6, p. 393-400. KUISLE, Oliver; QUIÑOÁ, Emilio and RIGUERA, Ricardo. A general methodology for automated solid-phase synthesis of depsides and depsipeptides. Preparation of a valinomycin analogue. Journal of Organic Chemistry, October 1999, vol. 64, no. 22, p. 8063-8075. [CrossRef] KULLMAN, Willi. Enzymatic synthesis of Leu- and Met-enkephalin. Biochemical and Biophysical Research Communications, November 1979, vol. 91, no. 2, p. 693-698. [CrossRef] KULLMAN, Willi. Protease-catalyzed peptide bond formation: application to synthesis of the COOH-terminal octapeptide fragment of cholecystokinin. Proceedings of the National Academy of Sciences of the United States of America, May 1982, vol. 79, no. 9, p. 2840-2844. [CrossRef] KUMAR, Dinesh and BHALLA, Tek Chand. Microbial proteases in peptide synthesis: approaches and applications. Applied Microbiology and Biotechnology, October 2005, vol. 68, no. 6, p. 726-736. [CrossRef] LAANE, C.; BOEREN, S.; HILHORST, R. and VEEGER, C. Optimization of biocatalysis in organic media. In: LAANE, C.; TRAMPER, J. and LILLY, M.D. eds. Biocatalysis in Organic Media. Elsevier Science Publishing Company, Inc., New York, 1987a, p. 65-84. LAANE, Colja; BOEREN, Sjef; VOS, Kees and VEEGER, Cees. Rules for optimization of biocatalysis in organic solvents. Biotechnology and Bioengineering, July 1987b, vol. 30, no. 1, p. 81-87. [CrossRef] LALONDE, J. Practical catalysis with enzyme crystals. Chemical Technology, February 1997, vol. 27, no. 2, p. 38-45. LEI, Han; WANG, Wei; CHEN, Ling-Li; LI, Xiao-Cong; YI, Bin and DENG, Le. The preparation and catalytically active characterization of papain immobilized on magnetic composite microspheres. Enzyme and Microbial Technology, July 2004, vol. 35, no. 1, p. 15-21. [CrossRef] LEVITSKY, Vladislav Yu; LOZANO, Pedro and IBORRA, José L. Kinetic analysis of deactivation of immobilized α-chymotrypsin by water-miscible organic solvent in kyotorphin synthesis. Biotechnology and Bioengineering, October 1999, vol. 65, no. 2, p. 170-175. [CrossRef] LIOY, Eduardo; SUAREZ, Jorge; GUZMAN, Fanny; SIEGRIST, Sibylle; PLUSCHKE, Gerd and PATARROYO, Manuel E. Synthesis, biological, and immunological properties of cyclic peptides from Plasmodium falciparum merozoite surface protein-1. Angewandte Chemie - International Edition in English, July 2001, vol. 40, no. 14, p. 2631-2635. [CrossRef] LIU, Chuan-Fa and TAM, James P. Subtilisin-catalyzed synthesis of amino acid and peptide esters. Application in a two-step enzymatic ligation strategy. Organic Letters, 2001, vol. 3, no. 26, p. 4157-4159. [CrossRef] LIU, Ping; TIAN, Gui-Ling; LEE, Kin-Sing; WONG, Man-Sau and YE, Yun-Hua. Full enzymatic synthesis of a precursor of bioactive pentapeptide OGP (10-14) in organic solvents. Tetrahedron Letters, March 2002, vol. 43, no. 13, p. 2423-2425. [CrossRef] LIU, Yu-Ying; LOU, Wen-Yong; ZONG, Min-Hua; XU, Ruo; HONG, Xiao and WU, Hong. Increased enantioselectivity in the enzymatic hydrolysis of amino acid esters in the ionic liquid 1-butyl-3methyl-imidazolium tetrafluoroborate. Biocatalysis and Biotransformation, March-April 2005, vol. 23, no. 2, p. 89-95. [CrossRef] LIU, Shu-Lai; WEI, Dong-Zhi; SONG, Qing-Xun; ZHANG, Ye-Wang; and WANG, Xue-Dong. Effect of organic cosolvent on kinetic resolution of tert-leucine by penicillin G acylase from Kluyvera citrophila. Bioprocess and Biosystems Engineering, April 2006, vol. 28, no. 5, p. 285-289. [CrossRef] LLOYD-WILLIAMS,
P. and GIRALT, E. Solid-phase convergent approaches to the synthesis
native peptides and proteins. In: KATES, LOBELL, Mario and SCHNEIDER, Manfred P. Pronase catalysed peptide syntheses. Journal of the Chemical Society - Perkin Transactions 1, January 1998, vol. 2, p. 319-326. [CrossRef] LOMBARD, C.; SAULNIER, J. and WALLACH, J. Recent trends in protease-catalyzed peptide synthesis. Protein and Peptide Letters, October 2005, vol. 12, no. 7, p. 621-629. LÓPEZ-SERRANO, P.; CAO, L.; VAN RANTWIJK, F. and SHELDON, R.A. Cross-linked enzyme aggregates with enhanced activity: application to lipases. Biotechnology Letters, August 2002, vol. 24, no. 16, p. 1379-1383. [CrossRef] LOU, Wen-Yong; ZONG, Min-Hua and WU, Hong. Enhanced activity, enantioselectivity and stability of papain in asymmetric hydrolysis of D,L-p-hydroxyphenylglycine methyl ester with ionic liquid. Biocatalysis and Biotransformation, May 2004, vol. 22, no. 3, p. 171-176. [CrossRef] LOZANO, Pedro; COMBES, Didier; IBORRA, José L. and MANJÓN, Arturo. Synthesis of L-Tyrosine glyceryl ester catalyzed by α-chymotrypsin in water-miscible organic solvents: a possible sun-tan accelerator product. Biotechnology Letters, December 1993, vol. 15, no. 12, p. 1223-1228. LOZANO, José Manuel; ESPEJO, Fabiola; OCAMPO, Marisol; SALAZAR, Luz May; TOVAR, Diana; BARRERA, Nubia; GUZMAN, Fanny and PATARROYO, Manuel Elkin. Mapping the anatomy of a Plasmodium falciparum MSP-1 epitope using pseudopeptide-induced mono- and polyclonal antibodies and CD and NMR conformation analysis. Journal of Structural Biology, October 2004, vol. 148, no. 1, p. 110-122. [CrossRef] LU, Yi-An and TAM, James P. Peptide ligation by a reversible and reusable C-terminal thiol handle. Organic Letters, October 2005, vol. 7, no. 22, p. 5003-5006. [CrossRef] MACHADO, Maria Fátima and SARAIVA, Jorge Manuel. Thermal stability and activity regain of horseradish peroxidase in aqueous mixtures of imidazolium-based ionic liquids. Biotechnology Letters, August 2005, vol. 27, no. 16, p.1233-1239. [CrossRef] MANABE, Kei; MORI, Yuichiro; WAKABAYASHI, Takeshi; NAGAYAMA, Satoshi and KOBAYASHI, Shü. Organic synthesis inside particles in water: lewis acid-surfactant-combined catalysts for organic reactions in water using colloidal dispersions as reaction media. Journal of the American Chemical Society, 2000, vol. 122, no. 30, p. 7202-7207. [CrossRef] MARTINEK, Karel and SEMENOV, Anatole N. Enzymatic synthesis in biphasic aqueous-organic systems. 2. Shift of ionic equilibria. Biochimica et Biophysica Acta, March 1981, vol. 658, no. 1, p. 90-101. [CrossRef] MARTINEK, Karel; SEMENOV, Anatole N. and BEREZIN, Ilya V. Enzymatic synthesis in biphasic aqueous-organic systems. 1. Chemical equilibrium shift. Biochimica et Biophysica Acta, March 1981, vol. 658, no. 1, p. 76-89. [CrossRef] MARTINEK, Karel; LEVASHOV, Andrey V.; KLYACHKO, Natalya; KHMELNITSKI, Yuri L. and BEREZIN, Ilya V. Micellar enzymology. European Journal of Biochemistry, March 1986, vol. 155, no. 3, p. 463-468. [CrossRef] MARTIN, S.L.; MCDOWELL, A.; LYNAS, J.F.; NELSON, J. and WALKER B. A study of the anti-invasive properties of N-α-phthalimidomethyl-ketomethylene tripeptide-based metalloprotease inhibitors. Journal of Pharmacy and Pharmacology, March 2001, vol. 53, no. 3, p. 333-343. [CrossRef] MERRIFIELD, Bruce. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. Journal of the American Chemical Society, July 1963, vol. 85, no. 14, p. 2149-2154. MERRIFIELD, Bruce. Solid phase synthesis. Science, April 1986, vol. 232, no. 4748, p. 341-347. [CrossRef] MERRIFIELD, Bruce. The chemical synthesis of proteins. Protein Science, September 1996, vol. 5, no. 9, p. 1947-1951. MESIANO, Anita J.; BECKMAN, Eric J. and RUSSELL, Alan J. Supercritical biocatalysis. Chemical Reviews, February 1999, vol. 99, no. 2, p. 623-634. [CrossRef] MIRANDA, Les P. and ALEWOOD, Paul F. Accelerated chemical synthesis of peptides and small proteins. Proceedings of the National Academy of Sciences of the United States of America, February 1999, vol. 96, no. 4, p. 1181-1186. [CrossRef] MIRANDA, Les P. and ALEWOOD, Paul F. Challenges for protein chemical synthesis in the 21st century: bridging genomics and proteomics. Biopolymers - Peptide Science, 2000, vol. 55, no. 3, p. 217-226. [CrossRef] MITIN, Y.V.; ZAPEVALOVA, N.P. and GORBUNOVA, E.Y. Peptide synthesis catalyzed by papain at alkaline pH values. International Journal of Peptide and Protein Research, March 1984, vol. 23, no. 5, p. 528-534. MITIN, Yuri V.; BRAUN, Karin and KUHL, Peter. Papain catalyzed synthesis of glyceryl esters of N-protected amino acids and peptides for the use in trypsin catalyzed peptide synthesis. Biotechnology and Bioengineering, May 1997, vol. 54, no. 3, p. 287-290. [CrossRef] MIYAZAWA, Toshifumi; NAKAJO, Shinichi; NISHIKAWA, Miyako; HAMAHARA, Kazumi; IMAGAWA, Kiwamu; ENSATSU, Eiichi; YANAGIHARA, Ryoji and YAMADA, Takashi. α-Chymotrypsin-catalysed peptide synthesis via the kinetically controlled approach using activated esters as acyl donors in organic solvents with low water content: incorporation of non-protein amino acids into peptides. Journal of the Chemical Society - Perkin Transactions 1, January 2001a, vol. 1, p. 82-86. [CrossRef] MIYAZAWA, Toshifumi; TANAKA, Kayoko; ENSATSU, Eiichi; YANAGIHARA, Ryoji and YAMADA, Takashi. Broadening of the substrate tolerance of α-chymotrypsin by using the carbamoylmethyl ester as an acyl donor in kinetically controlled peptide synthesis. Journal of the Chemical Society - Perkin Transactions 1, January 2001b, vol. 1, p. 87-94. [CrossRef] MONTER, B.; HERZOG, B.; STEHLE, P. and FÜRST, P. Kinetically controlled synthesis of dipeptides using ficin as biocatalyst. Biotechnology and Applied Biochemistry, October 1991, vol. 14, no. 2, p. 183-191. MOORE, William T. Laser desorption mass spectrometry. Methods in Enzymology, 1997, vol. 289, p. 520-542. [CrossRef] MORIHARA, Kazuyuki. Using protease in peptide synthesis. Trends in Biotechnology, June 1987, vol. 5, no. 6, p. 164-170. [CrossRef] MOZHAEV, Vadim V.; KHMELNITSKY, Yuri L.; SERGEEVA, Mariya V.; BELOVA, Alla B.; KLYACHKO, Nataliya L.; MARTINEK, Karel and LEVASHOV, Andrey V. Catalytic activity and denaturation of enzymes in water/organic cosolvent mixtures. Alpha-chymotrypsin and laccase in mixed water/alcohol, water/glycol and water/formamide solvents. European Journal of Biochemistry, October 1989, vol. 184, no. 3, p. 597-602. [CrossRef] MURAKAMI, Yoshihiko and HIRATA, Akira. New method for analysis of enzymatic reactions: thermolysin-catalyzed synthesis of aspartame precursor. Journal of Fermentation and Bioengineering, 1997, vol. 83, no. 1, p. 38-42. [CrossRef] MURAKAMI, Yoshihiko; YOSHIDA, Tatsunori; HAYASHI, Shigemitsu and HIRATA, Akira. Continuous enzymatic production of peptide precursor in aqueous/organic biphasic medium. Biotechnology and Bioengineering, July 2000, vol. 69, no. 1, p. 57-65. [CrossRef] MURPHY, A. and FAGAIN, C.O. Corrigendum dipeptide synthesis using acetylated trypsin derivative. Biotechnology Techniques, September 1997, vol. 11, no. 9, p. 707. [CrossRef] NAKANISHI, Kazuhiro and MATSUNO, Ryuichi. Kinetics of enzymatic synthesis of peptides in aqueous/organic biphasic system. Thermolysin-catalyzed synthesis of N-(benzyloxycarbonyl) -L-phenylalanine methyl ester. European Journal of Biochemistry, December 1986, vol. 161, no. 3, p. 533-540. [CrossRef] NAKANISHI,
Kazuhiro and MATSUNO, Ryuichi. Enzymatic synthesis of aspartame. In:
KING, R.D. and CHEETHAM, P.S.I. eds. Food Biotechnology. NAKANISHI, Kazuhiro; TAKEUCHI, Akira and MATSUNO, Ryuichi. Long-term continuous synthesis of aspartame precursor in a column reactor with an immobilized thermolysin. Applied Microbiology and Biotechnology, March 1990, vol. 32, no. 6, p. 633-636. [CrossRef] NARDIN, E.H.; CALVO-CALLE, J.M.; OLIVEIRA, G.A.; CLAVIJO, P.; NUSSENZWEIG, R.; SIMON, R.; ZENG, W. and ROSE, K. Plasmodium falciparum polyoximes: highly immunogenic synthetic vaccines constructed by chemoselective ligation of repeat B-cell epitopes and a universal T-cell epitope of CS protein. Vaccine, April 1998, vol. 16, no. 6, p. 590-600. [CrossRef] NILSSON, Bradley L.; SOELLNER, Matthew B. and RAINES, Ronald T. Chemical synthesis of proteins. Annual Reviews of Biophysics and Biomolecular Structure, June 2005, vol. 34, p. 91-118. [CrossRef] NISHIUCHI, Yuji; INUI, Tatsuya; NISHIO, Hideki; BODI, József; KIMURA, Terutoshi; TSUJI, Frederick I. and SAKAKIBARA, Shumpei. Chemical synthesis of the precursor molecule of the Aequorea green fluorescent protein, subsequent folding, and development of fluorescence. Proceedings of the National Academy of Sciences of the United States of America, November 1998, vol. 95, no. 23, p.13549-13554. [CrossRef] NOYA,
O.; OBREGÓN,
Walter D.; ARRIBÉRE, M. Cecilia; MORCELLE OGINO, Hiroyasu; UCHIHO, Takeshi; YOKOO, Jyunko; KOBAYASHI, Reina; ICHISE, Rikiya and ISHIKAWA, Haruo. Role of intermolecular disulfide bonds of the organic solvent-stable PST-01 protease in its organic solvent stability. Applied and Environmental Microbiology, February 2001, vol. 67, no. 2, p. 942-947. [CrossRef] OOSHIMA, Hiroshi; MORI, Hideki and HARANO, Yoshio. Synthesis of the aspartame precursor by solid thermolysin in organic solvent. Biotechnology Letters, November 1985, vol. 7, no. 11, p. 789-792. [CrossRef] OYAMA, Kiyotaka; IRINO, Shigeaki and HAGI, Norio. Production of aspartame by immobilized thermoase. Methods in Enzymology, 1987, vol. 136, p. 503-516. [CrossRef] PARK, Seongsoon and KAZLAUSKAS, Romas J. Biocatalysis in ionic liquids - advantages beyond green technology. Current Opinion in Biotechnology, August 2003, vol. 14, no. 4, p. 432-437. [CrossRef] PARTRIDGE, Johann; MOORE, Barry D. and HALLING, Peter J. α-Chymotrypsin stability in aqueous-acetonitrile mixtures: is the native enzyme thermodynamically or kinetically stable under low water conditions? Journal of Molecular Catalysis B: Enzymatic, January 1999, vol. 6, no. 1-2, p. 11-20. [CrossRef] PATARROYO, Manuel E.; AMADOR, Roberto; CLAVIJO, Pedro; MORENO, Alberto; GUZMAN, Fanny; ROMERO, Pedro; TASCON, Ricardo; FRANCO, Antonio; MURILLO, Luis A.; PONTON, Gabriel and TRUJILLO, Gustavo. A synthetic vaccine protects humans against challenge with asexual blood stages of Plasmodium falciparum malaria. Nature, March 1988, vol. 332, no. 6160, p. 158-161. [CrossRef] PATARROYO, J.H.; PORTELA, R.W.; DE CASTRO, R.O.; COUTO PIMENTELA, J.; GUZMÁN, F.; PATARROYO, M.E.; VARGAS, M.I.; PRATES, A.A. and DIAS MENDES, M.A. Immunization of cattle with synthetic peptides derived from the Boophilus microplus gut protein (Bm86). Veterinary Immunology and Immunopathology, September 2002, vol. 88, no. 3-4, p. 163-172. [CrossRef] PATARROYO,
J.H. and GUZMAN, F. Peptideos Sinteticos POGOREVC, Mateja and FABER, Kurt. Biocatalytic resolution of sterically hindered alcohols, carboxylic acids and esters containing fully substituted chiral centers by hydrolytic enzymes. Journal of Molecular Catalysis B: Enzymatic, September 2000, vol. 10, no. 4, p. 357-376. [CrossRef] PRIOLO, Nora; MORCELLE DEL VALLE, Susana; ARRIBÉRE, M. Cecilia; LÓPEZ, Laura and CAFFINI, Néstor. Isolation and characterization of a cysteine protease from the latex of Araujia hortorum fruits. Journal of Protein Chemistry, January 2000, vol. 19, no. 1, p. 39-49. [CrossRef] PRIOLO, Nora; ARRIBÉRE, M. Cecilia; CAFFINI, Néstor; BARBERIS, Sonia; NIETO VÁZQUEZ, Rodolfo and LUCO, Juan M. Isolation and purification of cysteine peptidases from the latex of Araujia hortorum fruits. Study of their esterase activities using partial least-squares (PLS) modeling. Journal of Molecular Catalysis B: Enzymatic, November 2001, vol. 15, no. 4-6, p.177-189. [CrossRef] QUIROGA, E.; NAVIGATORE FONZO, L. and BARBERIS, S. Catálisis enzimática en fase no acuosa. Parte I. Sistema de reacción. Anuario Latinoamericano de Educación Química ALDEQ, 2000a, vol. 13, p. 20-24. QUIROGA,
E.; BARBERIS, S.; ARRIBÉRE, M.C. and PRIOLO, N. Peptide enzymatic
synthesis in biphasic media. In: International Congress: 18th
Annual Scientific Meeting of Cuyo Biology Society (1st
December, 2000, Uspallata, QUIROGA, E.; PRIOLO, N.; MARCHESE, J. and BARBERIS, S. Stability of araujiain, a novel plant protease, in different organic systems. Acta Farmacéutica Bonaerense, April 2005, vol. 24, no. 2, p. 204-208. QUIROGA,
Evelina; PRIOLO, Nora; MARCHESE, José and BARBERIS, Sonia. Behaviour
of Araujiain, a new cysteine phytoprotease, in organic media
with low water content. Electronic Journal of RANCE, M.; SORENSEN, O.W.; BODENHAUSEN, G.; WAGNER, G.; ERNST, R.R. and WUTHRICH, K. Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochemical and Biophysical Research Communications, December 1983, vol. 117, no. 2, p. 479-485. [CrossRef] REETZ, Manfred T. and JAEGER, Karl-Erich. Enantioselective enzymes for organic synthesis created by directed evolution. Chemistry A European Journal, February 2000, vol. 6, no. 3, p. 407-412. [CrossRef] RESLOW, Mats; ADLERCREUTZ, Patrick and MATTIASON, Bo. Organic solvents for bioorganic synthesis. 1. Optimization of parameters for α-chymotripsin catalysed process. Applied Microbiology and Biotechnology, April 1987, vol. 26, no. 1, p. 1-8. [CrossRef] RINIKER, Bernhard; FLORSHEIMER, Andreas; FRETZ, Heinz; SIEBER, Peter and KAMBER, Bruno. A general strategy for the synthesis of large peptides: r1 the combined solid-phase and solution approach. Tetrahedron, 1993, vol. 49, no. 41, p. 9307-9320. [CrossRef] RINK, Hans. Solid-Phase synthesis of protected peptide fragments using a trialkoxy-diphenyl methylester resin. Tetrahedron Letters, 1987, vol. 28, no. 33, p. 3787-3790. [CrossRef] RIVERA, Z.; GRANADOS, G.; PINTO, M.; VARON, D.; CARVAJAL, C.; CHAVES, F.; CALVO, J.; RODRIGUEZ, R.; GUZMAN, F. and PATARROYO, M.E. Double dimer peptide constructs are immunogenic and protective against Plasmodium falciparum in the experimental Aotus monkey model. Journal of Peptide Research, February 2002, vol. 59, no. 2, p. 62-70. ROBERTS, Stanley M. Preparative biotransformations. Journal of the Chemical Society Perkin Transactions 1, 2001, vol. 14, p. 1475-1499. RODAKIEWICZ-NOWAK, J.; KASTURE, S.M.; DUDEK, B. and HABER, J. Effect of various water-miscibles solvents on enzymatic activity of fungal laccases. Journal of Molecular Catalysis B: Enzymatic, November 2000, vol. 11, no. 1, p. 1-11. [CrossRef] RODGER,
P.B.; DURRANT, ROIG, M. and KENNEDY, J. Perspectives for chemical modification of enzymes. Critical Reviews in Biotechnology, 1992, vol. 12, p. 391-412. ROMERO,
M.D.; AGUADO, J.; GUERRA, M.J.; ALVARO, G.; NAVARRO, R. and RUBIO,
E. Synthesis of delicious peptide fragments catalyzed by immobilized
proteases. In: BLASER, H.U.; BAIKER, A. and PRINS, R. eds. Studies
in Surface Science and Catalysis. Elsevier Science Publishers,
ROSAS, J.E.; HERNANDEZ, R.M.; GASCON, A.R.; IGARTUA, M.; GUZMAN, F.; PATARROYO, M.E. and PEDRAZ, J.L. Biodegradable PLGA microspheres as a delivery system for malaria synthetic peptide SPf66. Vaccine, August 2001, vol. 19, no. 31, p. 4445-4451. [CrossRef] RUIZ,
RUSSELL, A.J. and KLIBANOV, A.M. Inhibitor-induced enzyme activation in organic solvents. Journal of Biological Chemistry, August 1988, vol. 263, no. 24, p. 11624-11626. RYU, Keungarp and DORDICK, Jonathan S. How do organic solvents affect peroxidase structure and function? Biochemistry, March 1992, vol. 31, no. 9, p. 2588-2598. [CrossRef] SAKINA, K.; KAWAZURA, K.; MORIHARA, K. and YAJIMA, H. Enzymatic synthesis of cholecystokinin-octapeptide. Chemical and Pharmaceutical Bulletin, October 1988, vol. 36, no. 10, p. 3915-3919. SALLEH, A.B.; BASRI, M.; TAIB, M.; JASMANI, H.; RAHMAN, R., RAHMAN, M. and RAZAK, C. Modified enzymes for reactions in organic solvents. Applied Biochemistry and Bioengineering, October 2002, vol. 103, no. 1-3, p. 349-358. [CrossRef] SANCLIMENS, Glòria; CRESPO, Laia; GIRALT, Ernest; ALBERICIO, Fernando and ROYO, Miriam. Preparation of de novo globular proteins based on proline dendrimers. Journal of Organic Chemistry, August 2005, vol. 70, no. 16, p: 6274-6281. [CrossRef] SANTOVENA,
A.; OLIVA, A.; GUZMAN, F.; SARABIA, Francisco; CHAMMAA, Samy; SANCHEZ RUIZ, Antonio; ORTIZ, Laura Martin and LOPEZ HERRERA F. Jorge. Chemistry and biology of cyclic depsipeptides of medicinal and biological interest. Current Medicinal Chemistry, May 2004, vol. 11, no. 10, p. 1309-1332. SARIN, Virender K.; KENT, Stephen B.H.; TAM, James P. and MERRIFIELD, Robert B. Quantitative monitoring of solid-phase peptide synthesis by the ninhydrin reaction. Analytical Biochemistry, October 1981, vol. 117, no. 1, p. 147-157. [CrossRef] SARMENTO, Ana Cristina; SILVESTRE, Liliana; BARROS, Marlene and PIRES, Euclides. Cardosins A and B, two new enzymes available for peptide synthesis. Journal of Molecular Catalysis B: Enzymatic, September 1998, vol. 5, no. 1-5, p. 327-330. [CrossRef] SCHECHTER,
SCHELLENBERGER, Volker; JAKUBKE, Hans-Dieter; ZAPEVALOVA, Nina P. and MITIN, Yuri V. Protease-catalyzed peptide synthesis using inverse substrates: the influence of reaction conditions on the trypsin acyl transfer efficiency. Biotechnology and Bioengineering, June 1991, vol. 38, no. 1, p. 104-108. [CrossRef] SCHMID, A.; DORDICK, J.S.; HAUER, B.; KIENER, A.; WUBBOLTS, M. and WITHOLT, B. Industrial biocatalysis today and tomorrow. Nature, January 2001, vol. 409, no. 6817, p 258-268. [CrossRef] SCHREIBER, Jürg V and SEEBACH, Dieter. Solid phase synthesis of a β-dodecapeptide with seven functionalized side chains and cd-spectroscopic evidence for a dramatic structural switch when going from water to methanol solution. Helvetica Chimica Acta, December 2000, vol. 83, no. 12, p. 3139-3152. [CrossRef] SCHREIBER, Jürg V.; FRACKENPOHL, Jens; MOSER, Frank; FLEISCHMANN, Thomas; KOHLER, Hans-Peter E. and SEEBACH, Dieter. On the biodegradation of β-peptides. ChemBioChem, May 2002, vol. 3, no. 5, p. 424-432. [CrossRef] SERGEEVA, Maria V.; PARADKAR, Vikram M. and DORDICK, Jonathan S. Peptide synthesis using proteases dissolved in organic solvents. Enzyme and Microbial Technology, June 1997, vol. 20, no. 8, p. 623-628. [CrossRef] SERRALHEIRO, M.L.M.; PRAZERES, D.M.F. and CABRAL, J.M.S. Dipeptide synthesis and separation in a reversed micellar membrane reactor. Enzyme and Microbial Technology, December 1994, vol. 16, no. 12, p. 1064-1073. [CrossRef] SEWALD,
Norbert and JAKUBKE, Hans-Dieter. Peptides: Chemistry and Biology. SHAH, Shweta; SHARMA, Aparna and GUPTA, Munishwar N. Preparation of cross-linked enzyme aggregates by using bovine serum albumin as a proteic feeder. Analytical Biochemistry, April 2006, vol. 351, no. 2, p. 207-213. [CrossRef] SO, Jin-Eon; SHIN, Jong-Shik and KIM, Byung-Gee. Protease-catalyzed tripeptide (RGD) synthesis. Enzyme and Microbial Technology, February 2000, vol. 26, no. 2-4, p. 108-114. [CrossRef] ST. CLAIR, Nancy L. and NAVIA, Manuel A. Cross-linked enzyme crystals as robust biocatalysts. Journal of the American Chemical Society, 1992, vol. 114, no. 18, p. 7314-7316. [CrossRef] STEWART,
J.M. and YOUNG, J.D. Solid phase peptide synthesis. 2nd
ed. TAKAGI, Hiroshi; HIRAI, Kazunori; MAEDA, Yoshimi; MATSUZAWA, Hiroshi and NAKAMORI, Shigeru. Engineering subtilisin E for enhanced stability and activity in polar organic solvents. Journal of Biochemistry, April 2000, vol. 127, no. 4, p. 617-625. TAKAI,
H.; SAKATO, K.; NAKAMIZO, N. and ISOWA, Y. Enzymatic synthesis of
caerulein peptide. In: OYAMA, K. and NIIIMURA, S. eds. Peptide
Chemistry. Protein Research Foundation, TAM, J.P.; HEATH, W.F. and MERRIFIELD, B. An SN2 deprotection of synthetic peptides with a low concentration of HF acid in dimethyl sulfide: evidence and application in peptide synthesis. Journal of the American Chemical Society, 1983, vol. 105, no. 21, p. 6442-6456. TAM, James P. Synthetic peptide vaccine design: synthesis and properties of a high-density multiple antigenic peptide system. Proceedings of the National Academy of Sciences of the United States of America, August 1988, vol. 85, no. 15, p. 5409-5413. TAM, J.P.; LU, Y.; LIU, C.F. and SHAO, J. Peptide synthesis using unprotected peptides through orthogonal coupling methods. Proceedings of the National Academy of Sciences of the United States of America, December 1995, vol. 92, no. 26, p. 12485-12489. TAYLOR, C.K.; ABEL, P.V.; HULCE, M. and SMITH, D.D. Solvent effects on coupling yields during rapid solid-phase synthesis of CGRP (8-37) employing in situ neutralization. Journal of Peptide Research, January 2005, vol. 65, no. 1, p. 84-89. [CrossRef] THORMANN, Michael; THUST, Sven; HOFMANN, Hans-Jörg and BORDUSA, Frank. Protease-catalyzed hydrolysis of substrate mimetics (inverse substrates): A new approach reveals a new mechanism. Biochemistry, May 1999, vol. 38, no. 19, p. 6056-6062. [CrossRef] THUST, Sven and KOKSCH, Beate. Protease-catalysed peptide synthesis for the site-specific incorporation of alpha-fluoroalkyl amino acids into peptides. Journal of Organic Chemistry, March 2003, vol. 68, no. 6, p. 2290-2296. [CrossRef] TICKLER, Anna K.; BARROW, Colin J. and WADE, John D. Improved preparation of amyloid-beta peptides using DBU as Nalpha-Fmoc deprotection reagent. Journal of Peptide Science, September 2001, vol. 7, no. 9, p. 488-494. [CrossRef] TORRES, Sebastián and CASTRO, Guillermo R. Non-aqueous biocatalysis in homogeneous solvent systems. Food Technology and Biotechnology, October 2004, vol. 42, no. 4, p. 271-277. TRUSEK-HOLOWNIA, Anna. Synthesis of ZAlaPheOMe, the precursor of bitter dipeptide in the two-phase ethyl acetate-water system catalysed by thermolysin. Journal of Biotechnology, April 2003, vol. 102, no. 2, p. 153-163. [CrossRef] TUCHSCHERER, G. and MUTTER, M. Template assisted protein de novo design. Pure and Applied chemistry, November 1996, vol. 68, no. 11, p. 2153-2162. TYAGI, Renu and GUPTA, Munishwar. Stability of enzymes in water/organic co-solvent mixtures. Biotechnology Techniques, July 1998, vol. 12, no. 7, p. 569-570. [CrossRef] VABENO, Jon; LEJON, Tore; NIELSEN, Carsten Uhd; STEFFANSEN, Bente; CHEN, Weiging; OUYANG, Hui; BORCHARDT, Ronald T. and LUTHMAN, Kristina N. Phe-Gly dipeptidomimetics designed for the di-/tripeptide transporters PEPT1 and PEPT2: synthesis and biological investigations. Journal of Medical Chemistry, February 2004, vol. 47, no. 4, p. 1060-1069. [CrossRef] VAN BRUNT, Jennifer. Biosensors for bioprocesses. Bio/Technology, 1987, vol. 5, no. 5, p. 437. VAN RANTWIJK, Fred; MADEIRA LAU, Rute and SHELDON, Roger A. Biocatalytic transformations in ionic liquids. Trends in Biotechnology, March 2003, vol. 21, no. 3, p. 131-138. [CrossRef] VAN
UNEN, Dirk-Jan; ENGBERSEN, Johan F.J. and REINHOUDT, David N. Large
acceleration of α-chymotrypsin-catalyzed dipeptide formation
by 18-crown- VAN UNEN, Dirk-Jan; ENGBERSEN, Johan F.J. and REINHOUDT, David N. Studies on the mechanism of crown-ether-induced activation of enzymes in non-aqueous media. Journal of Molecular Catalysis B: Enzymatic, January 2001, vol. 11, no. 4-6, p. 877-882. [CrossRef] VAN UNEN, Dirk-Jan; ENGBERSEN, Johan F.J. and REINHOUDT, David N. Why do crown ethers activate enzymes in organic solvents? Biotechnology and Bioengineering, February 2002, vol. 77, no. 3, p. 248-255. [CrossRef] VOLKIN, David B.; STAUBLI, Andrea; LANGER, Robert and KLIBANOV, Alexander M. Enzyme thermoinactivation in anhydrous organic solvents. Biotechnology and Bioengineering, January 1991, vol. 37, no. 9, p. 843-853. [CrossRef] VON MOOS, Elisabeth and BEN, Robert N. Recent advances in the synthesis of C-linked glycoconjugates. Current Topics in Medical Chemistry, November 2005, vol. 5, no. 14, p. 1351-1361. [CrossRef] WALSH,
WERDER, Moritz; HAUSER, Helmut; ABELE, Stefan and SEEBACH, Dieter. β-Peptides as inhibitors of small-intestinal cholesterol and fat adsorption. Helvetica Chimica Acta, October 1999, vol. 82, no. 10, p. 1774-1783. [CrossRef] WESCOTT, Charles R. and KLIBANOV, Alexander M. The solvent dependence of enzyme specificity. Biochimica et Biophysica Acta, May 1994, vol. 1206, no. 1, p. 1-9. [CrossRef] WEST, J. Blair; HENNEN, William J.; LALONDE, James L.; BIBBS, Jeffrey A.; ZHONG, Ziyang; MEYER, Edgar F. and WONG, Chi Huey. Enzymes as synthetic catalysts: mechanistic and active-site considerations of natural and modified chymotrypsin. Journal of the American Chemical Society, 1990, vol. 112, no. 13, p. 5313-5320. [CrossRef] WHEELER, Carl J. and CROTEAU, Rodney. Terpene cyclase catalysis in organic solvent/minimal water media: demonstration and optimization of (+)-alpha-pinene cyclase activity. Archives of Biochemistry and Biophysics, July 1986, vol. 248, no. 1, p. 429-434. [CrossRef] WORLD HEALTH ORGANIZATION (WHO). Guidelines for the production and quality control of synthetic peptide vaccines. WHO Technical Report series, No. 889, 1999. WILSON, Lorena; ILLANES, Andrés; ABIÁN, Olga; PESSELA, Benevides C.C.; FERNÁNDEZ-LAFUENTE, Roberto and GUISÁN, José M. Co-aggregation of penicillin G acylase and polyionic polymers: an easy methodology to prepare enzyme biocatalysts stable in organic media. Biomacromolecules, May 2004a, vol. 5, no. 3, p. 852-857. [CrossRef] WILSON, Lorena; ILLANES, Andrés; PESSELA, Benevides; ABIÁN, Olga; FERNÁNDEZ-LAFUENTE, Roberto and GUISÁN, José M. Encapsulation of crosslinked penicillin G acylase aggregates in lentikats: evaluation of a novel biocatalyst in organic media. Biotechnology and Bioengineering, June 2004b, vol. 86, no. 5, p. 558-562. [CrossRef] WISEMAN,
A. Enzyme in therapy-theory and practice. In: WISEMAN, A. ed. Topics
in Enzyme and Fermentation Biotechnology 4. Ellis Horwood, WONG, Chi Huey; CHEN, Szu-Ta; HENNEN, William J; BIBBS, Jeffrey A.; WANG, Yu-Feng; LIU, Jennifer L.C.; PANTOLIANO, Michael W.; WHITLOW, Marc and BRYAN, Philip N. Enzymes in organic synthesis: use of subtilisin and a highly stable mutant derived from multiple site-specific mutations. Journal of the American Chemical Society, 1990, vol. 112, no. 3, p. 945-953. [CrossRef] WONG, Chi-Huey, SCHUSTER, Matthias, WANG, Peng and SEARS, Pamela. Enzymic synthesis of N- and O-linked glycopeptides. Journal of the American Chemical Society, 1993, vol. 115, no. 14, p. 5893-5901. [CrossRef] WONG, C.H. and WHITESIDES, G.M. Enzymes in Synthetic Organic Chemistry (Tetrahedron Organic Chemistry, Series 12). Pergamon Press, 1994, 388 p. ISBN 0-08-035941-8. WRIGHT, G.; CARVER, A.; COTTOM, D.; REEVES, D.; SCOTT, A.; SIMONS, P.; WILMUT, I.; GARNER, I. and COLMAN, A. High level expression of active human alpha-1-antitrypsin in the milk of transgenic sheep. Bio/Technology, 1991, vol. 9, no. 9, p. 830-834. [CrossRef] WUBBOLTS, Marcel G.; FAVRE-BULLE, Oliver and WITHHOLD, Bernard. Biosynthesis of synthons in two-liquid-phase media. Biotechnology and Bioengineering, October 1996, vol. 52, no. 2, p. 301-308. [CrossRef] XIANG, H.; XIANG, G.Y.; LU, Z.M.; GUO, L. and ECKSTEIN, H. Total enzymatic synthesis of cholecystokinin CCK-5. Amino Acids, August 2004, vol. 27, no. 1, p. 101-105. [CrossRef] YAN, Liang Z. and DAWSON, Philip E. Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. Journal of the American Chemical Society, January 2001, vol. 123, no. 4, p. 526-533. [CrossRef] YAN, Al-Xin; LI, Xuan-Wen and YE, Yun-Hua. Recent progress on immobilization of enzymes on molecular sieves for reactions in organic solvents. Applied Biochemistry and Biotechnology, May 2002, vol. 101, no. 2, p. 113-129. [CrossRef] YE, Yun-Hua; TIAN, Gui-Ling; XING, Guo-Wen; DAI, Dong-Cheng; CHEN, Gang and LI, Chong-Xi. Enzymatic synthesis of N-protected Leu-enkephalin and some oligopeptides in organic solvents. Tetrahedron, October 1998, vol. 54, no. 41, p. 12585-12596. [CrossRef] YIN, Hang; FREDERICK, Kendra K.; LIU, Dahui; WAND, A. Joshua and DEGRADO, William F. Arylamide derivatives as peptidomimetic inhibitors of calmodulin. Organic Letters, January 2006, vol. 8, no. 2, p. 223-225. [CrossRef] ZAKS,
Aleksey and KLIBANOV, Alexander M. Enzymatic catalysis in organic
media at ZAKS, Aleksey and KLIBANOV, Alexander M. Enzyme-catalyzed processes in organic solvents. Proceedings of the National Academy of Sciences of the United States of America, May 1985, vol. 82, no. 10, p. 3192-3196. ZAKS, Aleksey and KLIBANOV, Alexander M. Enzymatic catalysis in non-aqueous solvents. Journal of Biological Chemistry, March 1988a, vol. 263, no. 7, p. 3194-3201. ZAKS, Aleksey and KLIBANOV, Alexander M. The effect of water on enzyme action in organic media. Journal of Biological Chemistry, June 1988b, vol. 263, no. 17, p. 8017-8021. ZAKS, Aleksey and DODDS, David R. Application of biocatalysis and biotransformations to the synthesis of pharmaceuticals. Drug Discovery Today, December 1997, vol. 2, no. 12, p. 513-531. [CrossRef] ZHANG, Lianshan and TAM, James P. Lactone and lactam library synthesis by silver ion-assisted orthogonal cyclization of unprotected peptides. Journal of the American Chemical Society, April 1999, vol. 121, no. 14, p. 3311-3320. [CrossRef] Note: Electronic Journal of Biotechnology is not responsible if on-line references cited on manuscripts are not available any more after the date of publication. |
|
|